3.2. POLARIS - 2D Light Water Reactor Lattice Physics Module
Code Responsible(s): M. A. Jessee Contributors: M. A. Jessee, W. A. Wieselquist, A. M. Holcomb, S. W. Hart, J. W. Bae, K. S. Kim, C. A. Gentry, S. E. Skutnik, U. Mertyurek, J. Yang
ABSTRACT
Polaris is an easy-to-use light water reactor (LWR) lattice physics capability
for SCALE. Originally released in SCALE 6.2, Polaris uses the Embedded Self
Shielding Method (ESSM) for multigroup cross section processing and a transport
solver based on the Method of Characteristics (MOC). The ESSM computes
multigroup self-shielded cross sections using Bondarenko interpolation methods.
The background cross section used in the interpolation is determined by
iterative 2D MOC fixed-source transport calculations. Polaris is integrated with
ORIGEN for depletion calculations. Each pin-or each radial subregion of
the pin-is depleted based on the local power distribution. An optional
critical spectrum calculation is incorporated into the depletion
calculation and the output edits of few-group homogenized cross
sections. Few-group cross sections are archived to the lattice physics archive (.t16
and .x16 extension) file, which can be used in subsequent core simulator calculations.
Nuclide inventories are archived to the ORIGEN binary concentration file (.f71 extension),
which can be used in subsequent SCALE calculations for source term characterization or radition shielding.
Depletion and decay data are archived to the ORIGEN binary library file (.f33 extension), and can also be
used in subsequent source term calculations.
ACKNOWLEDGMENTS
The authors express gratitude to the U.S. Nuclear Regulatory Commission (NRC) for funding Polaris development. The author acknowledges D. Algama (former NRC), A. Bielen, L. Kyriazidis, and H. Esmaili for their support of the project.
3.2.1. Introduction
Polaris was introduced in SCALE 6.2 to provide 2D lattice physics analysis capability for light water reactor (LWR) fuel designs. For multigroup cross section processing, Polaris uses the Embedded Self-Shielding Method (ESSM) [POLARISWK12]. Unlike SCALE multigroup sequences that use XSProc, ESSM does not require user-defined unit cell definitions. ESSM computes multigroup cross sections using Bondarenko interpolation methods. The background cross section used in the interpolation is determined by iterative 2D fixed source transport calculations. Both the ESSM fixed-source calculations and the keff calculation utilize a Method of Characteristics (MoC) transport solver developed in the Exnihilo computational package.
Polaris is integrated with ORIGEN for depletion calculations. Each pin-or each radial subregion of the pin-is depleted based on the local power distribution. An optional critical spectrum calculation is incorporated into the depletion calculation for computing depletion reaction rates and/or few-group homogenized cross sections. A complete description of the Polaris computational methods is provided in [POLARISJWM+21].
Polaris provides an easy-to-use input format to allow users to set up lattice models with minimal lines of input. All recognized Polaris commands are shown in Table 3.2.1. Note that many commands support short and long forms. The allowed basic Types for input are described in Table 3.2.2. The special Polaris TYPES are shown in Table 3.2.3. The convention used in this manual is that basic types appear italicized and capitalized (Type), while special Polaris types appear in all caps (TYPE).
card |
long command |
short command(s) |
|---|---|---|
system |
system |
sys |
geometry |
geometry |
geom |
composition |
composition |
comp |
property |
property |
prop |
material |
material |
mat |
burnup |
bu or dbu |
- |
power |
power |
pow |
options |
option |
opt |
time |
t or dt |
- |
state |
state |
- |
branch block |
branch |
- |
pin geometry component |
pin |
- |
assembly pin map |
pinmap |
- |
assembly channel |
channel |
- |
assembly half gap |
hgap |
- |
channel box |
box |
- |
shield |
shield |
- |
deplete |
deplete |
- |
slab geometry component |
slab |
- |
power basis materials |
basis |
- |
assembly inserts |
insert |
- |
assembly control elements water cross geometry displacement maps spatial meshing detector tallies operating histories restart cumulative burnup |
control cross dxmap (dymap) mesh detector history bui (ti) |
- - - - - - - |
basic type |
description |
examples |
incorrect examples |
|---|---|---|---|
Word |
starts with a character A-Z or a-z and includes characters, numbers, underscores |
uox bor_water_500ppm FUEL |
uox_enr5.1 316SS uox-3.1 |
Int |
integer |
17 92235 2 565 |
31.4 uox |
Bool |
boolean/logical |
yes false |
TRUE No |
Real |
any number |
565 10.257 1.5e-6 |
yes bor_water |
String |
a single or double quoted string |
“INFMED” “Includes spaces” ‘NONE’ |
Includes spaces |
Value |
any non-word |
Int|Bool|Real| String |
Polaris type |
description |
variants |
|---|---|---|
STYPE |
system type |
PWR | BWR |
GTYPE |
geometry type |
ASSM | REFL |
CTYPE |
composition type |
NUM | WT | FORM | CONC | LW | UOX | ENRU | UN | USI |
PTYPE |
property type |
SOLP | DOPANT |
ETYPE |
control element type |
RODLET|BLADE |
OTYPE |
option type |
KEFF | BOND | ESSM | CRITS PEC | FG | DEPL | RUN | PRINT | GAMMA | GEOM | DATA |
The Polaris input supports a very flexible input scheme that allows some elements to be suppressed for better readability. With key=value type input, when the standard order of keys is used, the keys may be suppressed. Consider the following input specification as an example.
geometry GNAME : ASSM npins=Int ppitch=Real [sym=<FULL>|SE]
The geometry card requires a geometry name (GNAME) in the first group, then a geometry type (GTYPE) which is ASSM here indicating an assembly geometry. The remaining arguments have keys: “npins” with an integer value, “ppitch” with a real value, and the optional “sym” with either FULL or SE values (optional arguments are always shown in square brackets: [sym=<FULL>|SE]. The default value is in angle brackets: <FULL>. The pipe “|” shows an or relation (i.e., FULL or SE is an acceptable value). With the flexible input processing, the following inputs are all valid and identical.
geometry FuelNode : ASSM npins=15 ppitch=1.43 sym=FULL
geometry FuelNode : ASSM 15 1.43 FULL
geometry FuelNode : ASSM sym=FULL ppitch=1.43 npins=15
geometry FuelNode : ASSM 15 1.43
geometry FuelNode ASSM 15 1.43
The group separator “:” is suppressed in the last variant. This is possible in any situation where (1) the group is implicitly terminated by running out of arguments or (2) the next type does not match the expected type in the current group. For example, consider the hgap card:
hgap [ d ] [: M ]
In this card, d and M are values (without keys) defined as Real and material name (MNAME), respectively. The following form would automatically bypass the Real value, which allows a default, and set the interassembly gap material name as COOL.2.
hgap COOL.2
3.2.2. SCALE 6.3 Polaris Input Updates
For the release of SCALE 6.2.3, several new input cards were implemented into Polaris to model boiling water reactor (BWR) geometries and neutron/gamma detectors, which requires a gamma transport calculation. Moreover, improvements to existing input cards were implemented, along with the ability to specify time-dependent state properties and the ability to specify one or more depletion histories. This section describes the new and modified input cards that are included in the Polaris input format for SCALE 6.3, which are accessible as part of the release of SCALE 6.2.3.
To maximize backwards compatibility for input files developed with the original SCALE 6.2.0 release, the new and modified input cards are not available by default with SCALE 6.2.3. The new and modified input cards were activated if the input file begins with =polaris_6.3 rather than =polaris. The suffix “_6.3” is an indicator to the Polaris input processor to use the SCALE 6.3 input format. For the SCALE 6.3 release, the original input cards supported in the SCALE 6.2 input format will be available if the input file begins with =polaris_6.2, with the new SCALE 6.3 defaults being used for inputs with =polaris.
The new input cards to model BWR geometries include:
cross – define the interior water cross geometry of SVEA assembly designs;
dxmap (or dymap) – define displacement maps that indicate that translation of the pin center in the x- (or y-) direction;
control <BLADE> – define the control blade geometry;
mesh – define advanced spatial meshing options for different materials; and
option <GEOM> – define geometry tolerances, advance meshing options, and plotting options.
The modified input cards to model BWR geometries include:
pin – define circular and square-based geometry zones, as well as arbitrarily sized pins, e.g. size=1.5 water rod in some 9x9 BWR lattice designs; and
box – define channel box geometry with arbitrary number of zones and cutout regions.
For neutron/gamma detector modeling, there is a new detector card and an addition to the existing option <FG> card to enable output to the few-group cross section output (T16) file.
To control the gamma calculation, an option <GAMMA> has been added.
The new input cards for time-dependent modeling include:
history – define one or more operating histories in the input file; and
bui (or ti) – define restart cumulative burnup (or time) values.
The modified input cards for time-dependent modeling include:
state – define one or more time-independent or time-dependent state properties;
bu (or t) – define cumulative burnup (or time) values; and
dbu (or dt) – define incremental burnup (or time) values.
Example input files are included in the ${SCALE}/regression/input directory:
polaris.6.3.atrium9x9.inp and polaris.6.3.atrium10x10.inp – prototypic ATRIUM models;
polaris.6.3.blade1.inp and polaris.6.3.blade2.inp – control <BLADE> examples;
polaris.6.3.ge7x7.inp through polaris.6.3.ge10x10.inp – prototypic GE models;
polaris.6.3.svea100.inp and polaris.6.3.svea64.inp – prototypic SVEA models; and
polarisHistory.inp: history example.
3.2.3. Setup
The cards in this section generally appear at the beginning of an input file. Note that the manual is organized with each card starting a new page. This is especially convenient when printing a few cards across different sections.
3.2.3.1. title - case title lines
title Line1 Line2 … Linei … LineN
param |
type |
name |
details |
default |
Linei |
String |
line |
used in output file headers |
“DEFAULT TITLE” |
Examples:
title "Westinghouse 15x15"
title "Westinghouse 15x15"
"Condition: Hot Full Power"
"Date: 10/18/2012"
Comments:
The title card gives a title to this Polaris case, which appears as a descriptive header on the output file. The additional lines may be used to document a subcase or to embed additional information in the output file in an orderly way (e.g., author, date, project identifier).
The title card is optional.
See also:
lib
3.2.3.2. library - nuclear data libraries
lib [mg=*String*]
param |
type |
name |
details |
default |
mg |
String |
multigroup library |
multigroup cross section library |
“fine_n” |
Examples:
% a name of library in the DATA directory
% use SCALE 252g ENDF/B-VII.1 library
lib "fine_therm" % Alias for the V7.1 252g-neutron 47g-gamma library
% use SCALE 56g ENDF/B-VII.1 library
lib mg="broad_lwr" % Alias for the V7.1 56g-neutron 19g-gamma library
% a name of a local library in the temporary working directory
% (useful in SAMPLER calculations)
lib "perturbed_xs_library"
% fully specified path
lib "C:\scale6.3\data\scale.rev05.xn252v7.1"
Comments:
The lib card specifies the multigroup library location. See SCALE’s FileNameAliases.txt file in the installation directory for up-to-date library aliases for the fine and broad group libraries provided in SCALE’s data directory. Only the 252-group and the 56-group cross section libraries are recommended to be used in Polaris. Full specification of the file path is acceptable, as in the final example shown above.
The lib card is optional.
See also:
title
3.2.4. Geometry
The highest level structures in the model are named and defined with a geometry card. The general outline for a geometry definition is shown below. Two types of geometry are currently supported, ASSM for pressurized water reactor (PWR) or boiling water reactor (BWR) assemblies with fuel elements in a square-pitch, and REFL for an assembly-adjacent reflector.
geom GNAME : GTYPE arguments
param |
type |
name |
details |
default |
GNAME |
Word |
geometry name |
||
GTYPE |
- |
geometry type |
||
ASSM |
assembly |
see pin & pinmap |
||
REFL |
reflector |
see slab |
||
arguments |
- |
remaining arguments |
depends on GTYPE |
The control element geometry is also enumerated with types, as shown below. To model PWR-type rod cluster control assemblies (RCCAs), the RODLET element type is used in conjunction with pin definitions. In future releases of Polaris, other control element types, such as BWR-type control blades will be supported..
control INAME : ETYPE arguments
param |
type |
name |
details |
default |
INAME |
Word |
control element name |
||
ETYPE |
- |
control element type |
||
RODLET |
PWR-type RCCA |
requires PINIDs |
||
arguments |
- |
remaining arguments |
depends on ETYPE |
3.2.4.1. geometry<ASSM> – assembly
param |
type |
name |
details |
default |
GNAME |
Word |
assembly name |
||
GTYPE |
ASSM |
|||
npins |
Int |
number of pins |
on each side of the assembly |
|
ppitch |
Real |
pin pitch |
units: cm |
|
sym |
FULL|SE |
symmetry |
assembly symmetry, FULL: no symmetry, SE: south-east quarter |
FULL |
modular |
Bool |
modular option |
True: modular geometry False: standard geometry Requires sym=FULL |
False |
Examples:
% simple pincell
geom MyPin : ASSM 1 1.5
% 17x17 Westinghouse with 1.26 cm pin pitch in quarter symmetry
geom FuelNode : ASSM 17 1.26 sym=SE
Comments:
Examples:
% simple pincell
geom MyPin : ASSM 1 1.5
% 17x17 Westinghouse with 1.26 cm pin pitch in quarter symmetry
geom FuelNode : ASSM 17 1.26 sym=SE
% Large 200x200 map of pins using modular option
geom FuelNode : ASSM 200 1.5 sym=FULL modular=True
Comments:
The assembly geometry describes the basic elements of an assembly. The pin and pinmap cards are required to finalize the assembly geometry. The hgap card specifies the interassembly half gap, and the channel specifies the channel material for the assembly.
The modular option is useful for large arrays of pins—for example, critical benchmarks such as KRITZ, DIMPLE, BW 1810. This option should be used only for problems >35 pins per side, FULL symmetry only, and without interassembly gaps or channel boxes. The modular option is set to False (or no) by default.
See also:
pinmap, pin, hgap, box, channel, control, insert
3.2.4.2. geometry<REFL> - reflector
param |
type |
name |
details |
default |
GNAME |
Word |
reflector name |
||
GTYPE |
REFL |
|||
thick |
Real |
thickness |
units: cm |
Examples:
% defines a 20 cm reflector
geom ReflectorNode : REFL 20.0
Comments:
The reflector geometry describes the basic elements of a simple slab-type reflector. The slab card can be used to define geometric dimensions and mesh for the reflector geometry.
See also:
slab
3.2.4.3. channel - coolant channel
channel [Mchan =MCLASS]
param |
type |
name |
details |
default |
Mchan |
MCLASS |
material class |
initializes materials in outermost pin zone |
* |
*By default, Mchan will be set to COOL by “system PWR” and “system BWR.” Otherwise, Mchan is required. |
Examples:
% define the channel material class to be COOL
channel COOL
Comments:
The channel card is used to set the default channel material class for the outermost region of each pin, typically containing reactor coolant. See the material card for a description of material classes.
See also:
pin, material, geometry<ASSM>
3.2.4.4. hgap - half distance between assemblies
param |
type |
name |
details |
default |
di |
Real |
list of widths with i: E: east N: north W: west S: south |
accepts 1, 2, or 4 values E: all hgaps are same E+N: dE =d:sub: S and dN =dW units: cm |
0.0 |
Mi |
MNAME |
list of material names |
requires same # as di |
* |
meshing options |
||||
nfi |
Int |
list of number of faces per pin |
requires same # as di |
2 |
ndi |
Int |
list of number of divisions |
requires same # as di |
1 |
*By default, hgap material will be set to COOL.1 by “system PWR.” For “system BWR,” the east and south hgap materials will be set to MOD.2, and the west and north hgap materials will be set to MOD.1. Otherwise hgap material is required.
Examples:
% defines a 17x17 Westinghouse assembly with 1.26 cm pin pitch
% with 0.04 cm half-gap filled with material COOL.1
geom w17x17 : ASSM 17 1.26 sym=SE
hgap 0.04 COOL.1
% defines a GE 7x7 assembly with 1.88 cm pin pitch
% 0.48 cm narrow gap on east and south edge
% 0.95 cm wide gap on north and west edge
% narrow gap mesh is 3
% wide gap mesh is 4
% faces per pin is 2 for both narrow and wide gap
geom ge7x7 : ASSM 7 1.88
hgap 0.48 0.95 : MOD.1 MOD.1 : 2 2 : 3 4
Comments:
The hgap specifies the outermost geometry region in an assembly. If a channel box exists, then hgap specifies the material and mesh from outer channel box edge to the problem boundary. Otherwise, hgap specifies the material and mesh from the edge of the fuel array to the problem boundary. In both cases, hgap represents the half-distance between adjacent assemblies for single assembly calculations. Fig. 3.2.1 shows some of the hgap meshing options. Referring to the south edge of the assembly, the number of faces per pin refers to the extra cells introduced by “splitting” the pin cell boundary, and the number of divisions refers to extra horizontal lines dividing half gap into smaller width cells.
See also:
pinmap, control, insert, geometry<ASSM>, channel, box
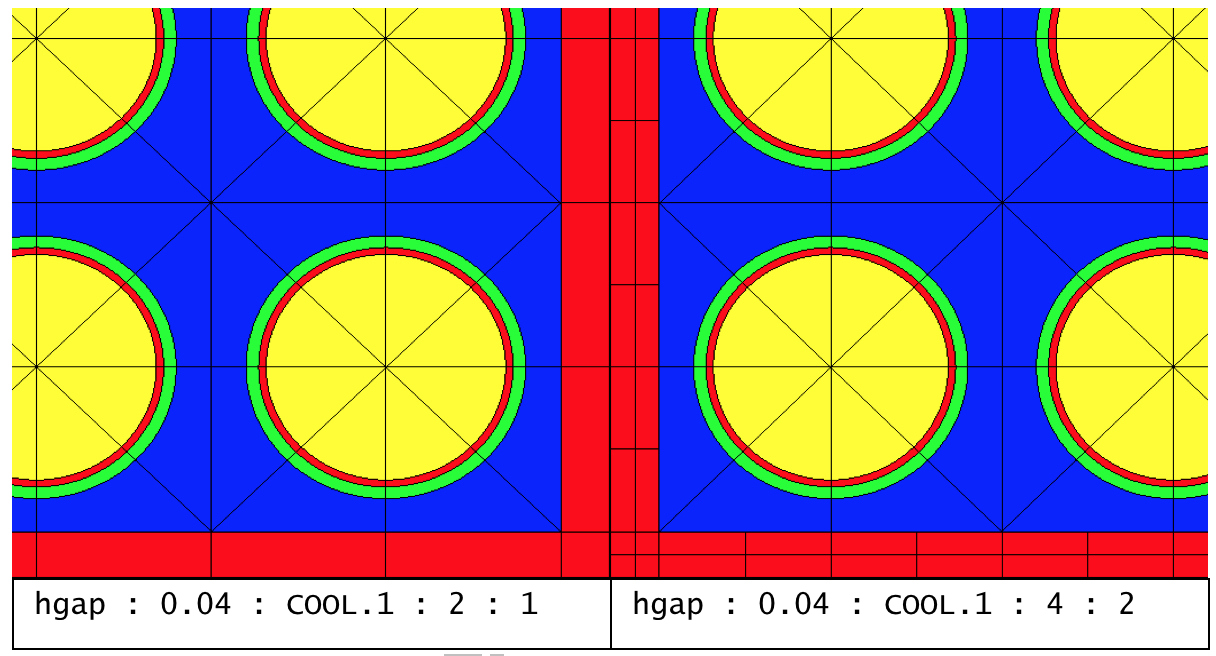
Fig. 3.2.1 Interassembly half gap meshing variants.
3.2.4.5. box - channel box geometry
box thick=Real [rad=*Real*] [hspan=*Real*] [Mbox=MNAME] [cothick=*Real*] [cobtm=*Real*] [cotop=*Real*]
param |
type |
name |
details |
default |
thick |
Real |
nominal thickness (cm) |
must be > 0.0 |
|
rad |
Real |
inner corner radius (cm) |
must be \(\geq\) 0.0 |
0.0 |
hspan |
Real |
half inner span (cm) |
——-See comments— —- |
|
Mbox |
MNAME |
box material |
* |
|
cothick |
Real |
interior cutout region thickness (cm) |
thick > cothick \(\geq\) 0.0 |
0.0 |
cobtm |
Real |
distance from box centerline to bottom of interior cutout region (cm) |
hspan-rad > cobtm \(\geq\) 0.0 |
0.0 |
cotop |
Real |
distance from box centerline to top of interior cutout region (cm) |
cobtm \(\geq\) cotop \(\geq\) 0.0 |
cobtm |
options for additional box zones |
||||
ti |
Real |
zone thickness (cm) |
must be \(\geq\) 0 |
|
ai |
Real |
distance from box centerline to bottom of zone cutout region (cm) |
—–See comments— – |
|
bi |
Real |
distance from box centerline to top of zone cutout region (cm) |
—–See comments— – |
|
Mi |
MNAME |
zone material |
—–See comments— – |
|
ri |
Real |
zone inner corner radius (cm) |
—–See comments— – |
*By default, box material will be set to CAN.1 by “system BWR.” Otherwise box material is required.
Examples:
% simple
box 0.2
% rounded corner, rad 0.9
box 0.2 0.9
% rounded corner and user-defined inner span
box 0.2 0.9 6.7
% two zones
box 0.2 0.9 6.7
: 0.2
: 4.0
: 4.3
Comments:
The box specifies the channel box geometry that surrounds the pinmap. The three primary dimensions of the channel box are the thickness (thick), the inner corner radius (rad), and the half inner span (hspan). Several additional dimensions for both box and cross are defined with respect to the channel box center. The channel box center is not to be confused with the lattice center: the former is the centroid of the inner channel box square boundary and the latter will depend on the wide and narrow gap dimensions provided on the hgap card. By default, the half inner span is equal to the half pin pitch multiplied by the number of pins on each side of the assembly (see npins and ppitch on the geometry<ASSM> card). If a cross card is applied, the default half inner span is increased by the half width of the interior cross buffer region (see hwidth on the cross card).
Additional channel box zones can be specified on the box card. The additional zones are useful for defining thick corner regions of the channel box. Each additional zone must have a user-defined thickness (ti, i = 2 to N). Note that the starting index begins at “2” rather than “1” because the zone 1 thickness has already been defined by the “thick” input field.
“Cutout regions” may be defined in which a portion of the channel box zone is replaced by the corresponding hgap material along the horizontal and vertical centerlines of the channel box. The cutout region is defined by the distance from the channel box centerline to the bottom additional channel box zone (ai) and the top of the channel box zone (bi). The values of ai and bi determine the size of trapezoidal cutout region centered along each face of the channel box. The bi value must be greater than or equal to the ai value. The ai value must be greater than or equal to the previous zone’s bi value, i.e., bi-1. By default, a2 and b2 are zero. If only M cutout regions are specified for N additional zones, i.e., M < N, both ai and bi is set to bM for i = M+1 to N.
Additional zones can also have a different inner corner radius (r2 … rN). The outer corner radius of the last zone may also be specified (rN+1). By default, r2 is zero if rad is zero. If rad is greater than zero, the default value of r2 is rad+thick. Similar rules apply for determining the default corner radii for additional zones if they are omitted in the input specification.
Additional zones can also have a different material (Mi). By default, M2 is Mbox. If additional materials are omitted in the input, the default value of Mi is Mi-1 for i = 3 to N.
The spatial mesh along each face of the channel box will be determined by the nf values specified on the hgap card.
The four examples listed above are displayed in Fig. 3.2.2. For additional examples, see the polaris.6.3 regression input files described at the beginning of Sect. 3.2.2.
See also:
geometry<ASSM>, hgap, cross
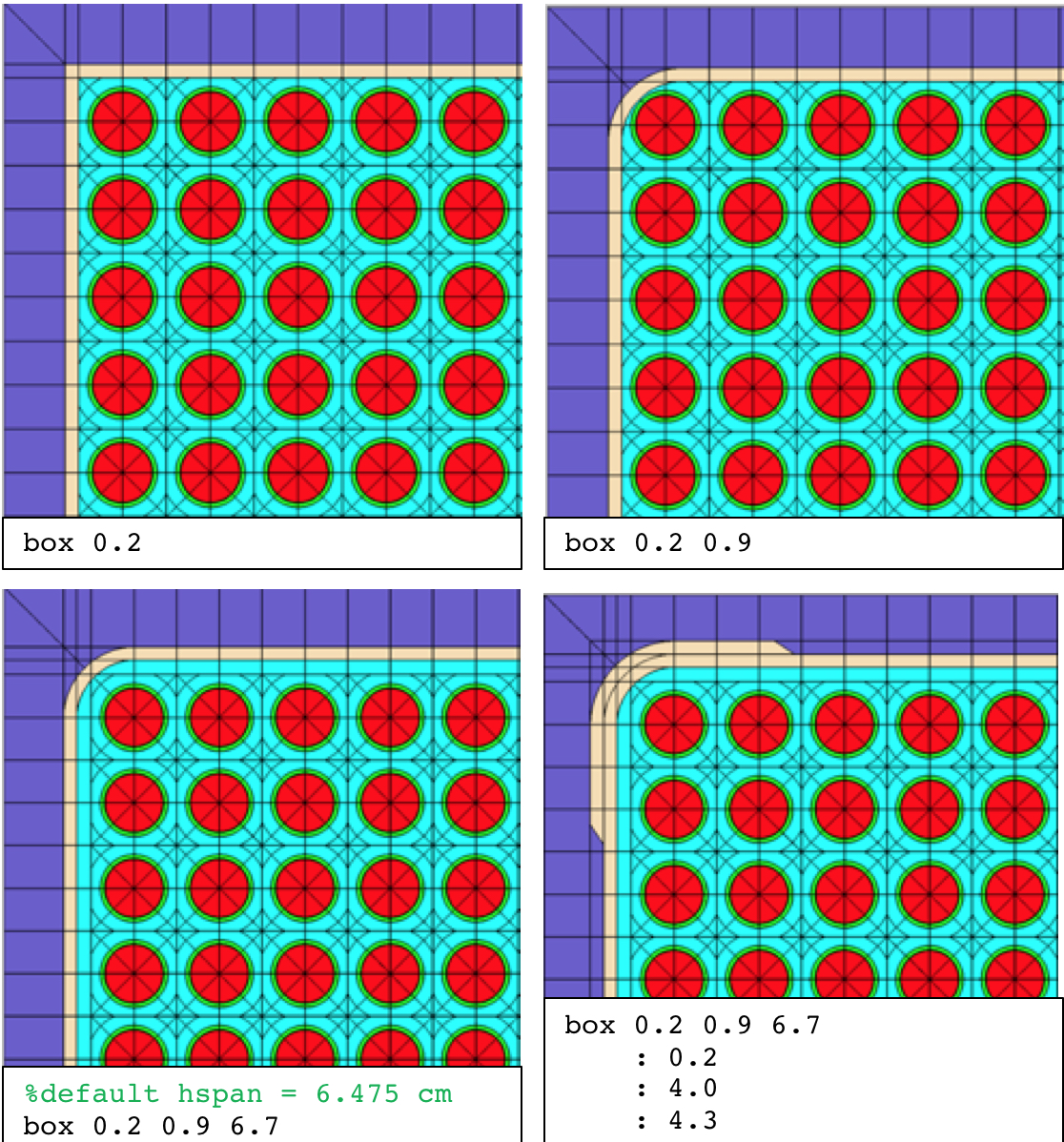
Fig. 3.2.2 Box card examples.
3.2.4.6. pin - pincell comprised of nested geometry zones of variable shape
param |
type |
name |
details |
default |
PINID |
Word|Int |
pin identifier |
||
size |
Real |
pin size multiplicat ion factor |
must be \(\geq\) 1.0 |
1.0 |
ri |
Real |
zone interior radius (cm) |
r1 must be > 0. Additional zones must be > ri-1 |
|
Mi |
MNAME |
zone material |
.1 added if given MCLASS, e.g., FUELFUEL.1 |
|
Mout |
MNAME |
outermost zone material |
.1 added if given MCLASS |
* |
Si |
CIR or SQR or SQR(X) |
circular zone or square zone with optional corner radius, X \(\geq\) 0.0 |
CIR X=0.0 |
*If not specified, the material class MCLASS is taken from the channel card (Mchan) and set to the first member of that class, “Mchan.1.” For example if Mchan=”COOL,” then Mout= “COOL.1.”
Examples:
%standard fuel pin
pin 1 : 0.4096 0.418 0.475 : FUEL.1 GAP.1 CLAD.1
%2x2 water rod
pin W 2.0 : 1.6 1.7 : MOD.1 TUBE.1
%3x3 square water box (ATRIUM)
pin W 3.0 : 1.68 1.75 : MOD.1 TUBE.1 : SQR SQR
%noninteger size water rod (GE9x9)
pin W 1.76 : 1.16 1.259 : MOD.1 TUBE.1 COOL.2
Comments:
The pin card is one of the basic building blocks of the assembly model. pin and slab are the only geometry components which allows an integer (Int) identifier as well as a Word-all other geometric identifiers use Word. Note that the materials are required, except for the last Mout, which can be used to overwrite the material given by a channel for the outermost region in the pincell. The pin geometry is constructed from the inside out, using either circle zones (defined by the radius) or square zones (defined by the half-width, and optional corner radius). Different examples of pin geometries are displayed in Fig. 3.2.3. All meshing options for the pin are provided through the mesh card.
If the pin size is an integer value, the pin consumes a size \(\times\) size subarray in the pinmap (e.g. 1 \(\times\) 1, 2 \(\times\)times` 3, etc). If the pin size is noninteger, the pin consumes a ceil(size) \(\times\) ceil(size) subarray in the pinmap. ceil(x) represents the ceiling function to round the value of x to the nearest integer greater than or equal to x. For size equal to 1.3, each instance of the pin will consume a 2 \(\times\) 2 subarray in the pinmap. Each instance of a noninteger-sized pin must share a location with another instance of a noninteger-sized pin, but not necessarily the same pin. The shared location must be set to “_” in the pinmap. The identification of the shared location is necessary to determine the center of each pin. The pin center is at a distance of size*half pitch*sqrt(2) from the opposite corner of the shared location, along the diagonal of the pin boundary. An example of an integer-sized pins is displayed in Fig. 3.2.3. An example of noninteger-sized pins is displayed in Fig. 3.2.4.
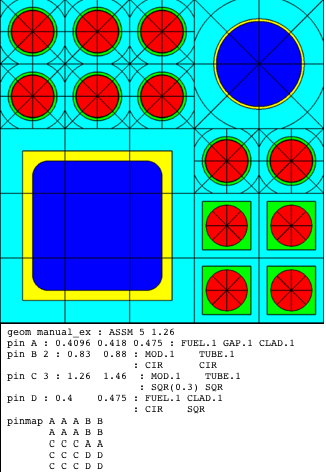
Fig. 3.2.3 Pin examples with different shape geometries
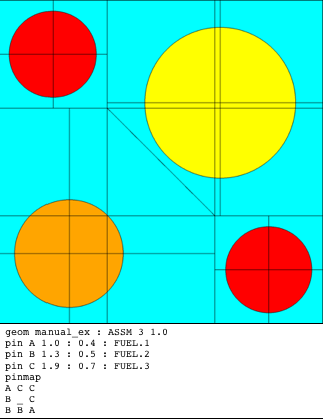
Fig. 3.2.4 Pin examples with noninteger pin size.
For additional examples, see the polaris.6.3 regression input files described at the beginning of Sect. 3.2.2
See also:
slab, pinmap, channel, mesh
3.2.4.7. pinmap - pin layout
param |
type |
name |
details |
default |
PINIDi |
Word|Int |
list of pin identifiers |
supports full, quarter, or octant symmetry quarter: assumes southeast (SE) octant: assumes south-by-so utheast (SSE) |
Examples:
%Westinghouse 17x17 pinmap in octant symmetry
pinmap
2
1 1
1 1 1
3 1 1 3
1 1 1 1 1
1 1 1 1 1 3
3 1 1 3 1 1 1
1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1
%Westinghouse 17x17 pinmap in quarter symmetry
pinmap
2 1 1 3 1 1 3 1 1
1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1
3 1 1 3 1 1 3 1 1
1 1 1 1 1 1 1 1 1
1 1 1 1 1 3 1 1 1
3 1 1 3 1 1 1 1 1
1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1
%Westinghouse 17x17 pinmap in full
pinmap
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 3 1 1 3 1 1 3 1 1 1 1 1
1 1 1 3 1 1 1 1 1 1 1 1 1 3 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 3 1 1 3 1 1 3 1 1 3 1 1 3 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 3 1 1 3 1 1 2 1 1 3 1 1 3 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 3 1 1 3 1 1 3 1 1 3 1 1 3 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 3 1 1 1 1 1 1 1 1 1 3 1 1 1
1 1 1 1 1 3 1 1 3 1 1 3 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
%large central 2x2 water rod in 6x6 assembly
%pinmap must show adjacent Ws
pin W size=2 : 0.8
: COOL
pinmap
F F F F F F
F F F F F F
F F W W F F
F F W W F F
F F F F F F
F F F F F F
Comments:
The pinmap card defines the layout of pin cells in the assembly. The symmetry is determined by the number of pin identifiers given on the card and must not be more general than the symmetry option given on the assembly geometry card (i.e., do not define a full pin map for a sym=SE assembly model). If the pin has a large size specifier, size>1, then the pinmap must reflect that with those pins occurring in blocks of size \(\times\) size.
See also:
pin, control, insert
3.2.4.8. control<RODLET> - RCCA-type layout
param |
type |
name |
details |
default |
INAME |
Word |
insert name |
||
ETYPE |
RODLET |
|||
PINIDi |
Word|Int |
list of pin identifiers |
same format as pinmap “_” indicate empty locations |
Examples:
% B4C control rods
mat GAS.1 : FILLGAS
mat CLAD.1 : ZIRC4
mat MOD.1 : LW
mat TUBE.1 : SS304
mat CNTL.1 : B4C
pin B : 0.214 0.231 0.241 0.427 0.437 0.484 0.561 0.602
: GAS TUBE GAS CNTL GAS TUBE MOD CLAD
control BankD : RODLET
_
_ _
_ _ _
B _ _ B
_ _ _ _ _
_ _ _ _ _ B
B _ _ B _ _ _
_ _ _ _ _ _ _ _
_ _ _ _ _ _ _ _ _
Comments:
Note that control elements and inserts share the INAME identifiers, so an insert and a control element cannot have the same name. Different control rod banks may be included in a single input file using more than one control card with unique INAMEs. The main difference between the inserts defined by control element and insert cards is that by default, control element materials are not depleted, whereas insert materials are depleted.
The outer dimensions of the tube must be included in the pin card that is inserted.
See also:
pinmap, control, insert, state
3.2.4.9. control<BLADE> - BWR control blade
param |
type |
name |
details |
default |
hwgthck |
Real |
half blade wing thickness (cm) |
must be >0 |
|
sththck |
Real |
sheath thickness (cm) |
must be >=0 |
|
cslnth |
Real |
central support length (cm) |
must be >=hwgthck |
|
sthmat |
MNAME |
sheath material |
STRUCT.1* |
|
csmat |
MNAME |
central support material |
STRUCT.1* |
|
hcsthck |
Real |
half central support thickness (cm) |
must be >0 |
hwgthck |
wgcrv |
Real |
wing tip radius (cm) |
must be >=0 |
0 |
IDi ` |
Word|Int |
pin or slab identifier |
——- See comments ———- |
|
Li |
Real |
length of section i |
——- See comments ———- |
|
Ni |
Real |
# of pins or slabs in section i |
——- See comments ———- |
|
*Default values for shtmat and csmat are set by “system BWR.” If “system BWR” is omitted, the material definitions are required. |
Comments:
The blade card defines a control blade geometry. The control blade identifier (INAME) can be used to insert the control blade using state or add statements to define histories or branches respectively. INAME=yes inserts the control blade into the northwest corner of the lattice.
The control blade geometry is described with reference to the blade wing on the northern edge of the lattice in Fig. 3.2.5. The blade wing on the west edge is a reflection of the northern edge wing along the diagonal symmetry line that extends from the northwest corner to the southeast corner of the lattice.
The two primary regions of the blade are the central support and the active blade wing. The central support has a length (cslnth), half width (hcsthck), and material (csmat). The central support half width is the vertical distance between the north face of the lattice and the south boundary of the central support. The central support length is the horizontal distance from west face of the lattice to the east boundary of the central support. See Fig. 3.2.5 for details.
The active portion of the blade wing begins at the east boundary of the central support. The active portion has a half width (hwgthck), sheath thickness (sththck), sheath material (sthmat), and wing tip radius (wgcrv). The half width is the vertical distance between the north face of the lattice and the southern boundary of the active blade wing, including the sheath. The wing tip radius can be any nonnegative number. If the radius is zero, the wing tip is a straight edge.
The active portion of the blade wing is subdivided into sections. Each section has a length (Li), and identifier associated with a pin or slab (IDi), and the number of pins or slabs for each section (Ni). The list of section lengths and section identifiers is required and must have consistent list lengths. The final list for number of pin/slabs per section is optional. If omitted, the default number of pin or slabs per section is one.
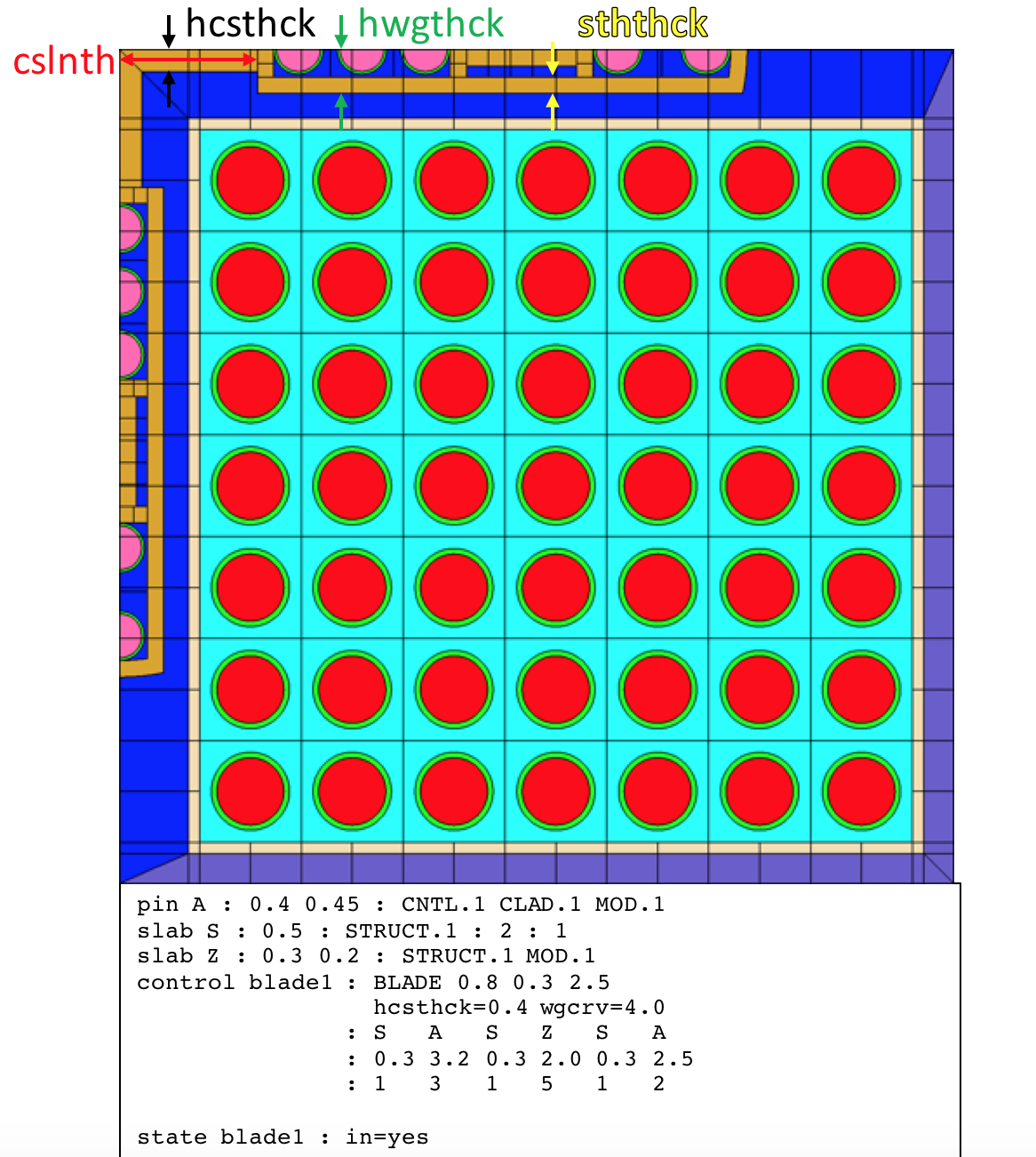
Fig. 3.2.5 Control blade example.
If there is only one pin in a pin section, the pin is placed in the section center. If there are multiple pins, the first and last pin are positioned flush against the west and east section boundary respectively, and the interior pins are uniformly spaced between the two edge pins. Slab sections are built from the blade centerline in the vertical direction towards the interior sheath boundary. Each slab zone has width equal to the section length. Each slab zone can be subdivided in the vertical direction by the zone nx parameter on the slab card. The slab zone can be subdivided in the horizontal direction by the product of the zone ny parameter and the number of slabs in the blade wing section.
The blade is further subdivided by the mesh nf/nd settings for hgap material in the north and west bypass region, typically MOD.1 for models that include system BWR.
See also: pin, slab, mesh, system BWR
3.2.4.10. insert - insert layout
param |
type |
name |
details |
default |
INAME |
Word |
insert name |
||
PINIDi |
Word|Int |
list of pin identifiers |
same format as pinmap “_” indicate empty locations |
Examples:
%pyrex inserts
pin P : 0.214 0.231 0.241 0.427 0.437 0.484 0.561 0.602
: GAS TUBE GAS BP.3 GAS TUBE COOL CLAD
insert PyrexInserts :
_
_ _
_ _ _
P _ _ P
_ _ _ _ _
_ _ _ _ _ P
P _ _ P _ _ _
_ _ _ _ _ _ _ _
_ _ _ _ _ _ _ _ _
Comments:
The insert card defines a set of pins to be used to model inserts such as WABA. When the insert is “in,” the insert pins replace overlapping regions of the pins defined on the assembly pinmap. An underscore (_) is used to indicate locations without inserts. See the notes on the control<RODLET> card for additional guidelines.
See also:
pinmap, control, insert
3.2.4.11. slab - slab geometry
param |
type |
name |
details |
default |
SLABID |
Word |
slab geometry identifier |
reflector GNAME |
|
ti |
Real |
list of slab thicknesses |
units: cm |
|
Mi |
MNAME |
list of material names |
||
meshing options |
||||
nxi |
Int |
list of number of x-divisions |
1 |
|
nyi |
Int |
list of number of y-divisions |
1 |
Examples:
% a reflector definition
% 2.22 cm of baffle
% 15 cm of moderator
geom ReflectorNode : REFL 17.22
slab 2.22 15 : BAFFLE.1 MOD.1
Comments:
The slab card may be used to define three things: (1) the materials and thicknesses of a reflector initiated on a geometry card, (2) slabs in a control blade, and (3) spacer grids. If the first argument identifier is not present, then the first purpose of describing the various material thicknesses in a reflector is assumed. The meshing options allow each material slab to be spatially refined in x and y, increasing the number of cells in the transport problem. The meshing option for the number of x divisions creates the equivalent of additional “sub-slabs” in each user-defined slab thickness. The y-divisions create additional cells vertically. The default of one y-division corresponds to the entire assembly.
See also:
geometry<REFL>
3.2.4.12. cross - cross geometry

|
Comments:
The cross card performs two tasks. First, it subdivides the pinmap into four subarrays, optionally adding a horizontal and vertical gap between the subarrays. The row parameter is uses to subdivide the pinmap. If the pinmap is 10 \(\times\) 10 and row=5, each of the four subarrays is 5 \(\times\) 5. If the pinmap is 10 \(\times\) 10 and row=4, the northwest subarray is 4 \(\times\)times`6, the southwest subarray is 6 \(\times\) 4, and the southeast subarray is 6 \(\times\) 6. The hwidth parameter controls the half-spacing of the horizontal and vertical gap in between the subarrays. The hwidth parameter must be \(\geq\) 0.0 and if hwidth is > 0.0, the gap is filled with material Mout (default is COOL.2 with system BWR).
The second task is the insertion of the cross structure into the lattice geometry. The process is described with reference to the example in Fig. Fig. 3.2.6. In the example, the pinmap is 9 \(\times\) 9 and row=3, hwidth=1.5, and hspan=10.5. The top left plot contains the four following lines:
the line in the center of the vertical cross gap,
the line in the center of the horizontal cross gap,
the diagonal line from the northwest (NW) channel box corner to the southeast (SE) corner, and
the diagonal line, perpendicular to line 3, passing through the intersection of line 1 and line 2.
These four lines intersect and form 8 separate regions, i.e., octants, within the channel box interior. The intersection point, i.e., cross center, is not necessarily equal to the box center as shown in this example. In the top left plot, the red triangle represents the WNW octant. In the bottom left plot, the red triangle represents the SSE octant.
% Centered cruciform flow channel
cross 0.625 0.08 5
: -6.86 -6.53 -5.20 -4.81 -3.77 -3.39 -2.05 0.0 2.05 3.39 3.77 4.81 5.20 6.53 6.86
: 0.625 0.0 0.0 0.24 0.24 0.0 0.0 2.05 0.0 0.0 0.24 0.24 0.0 0.0 0.625
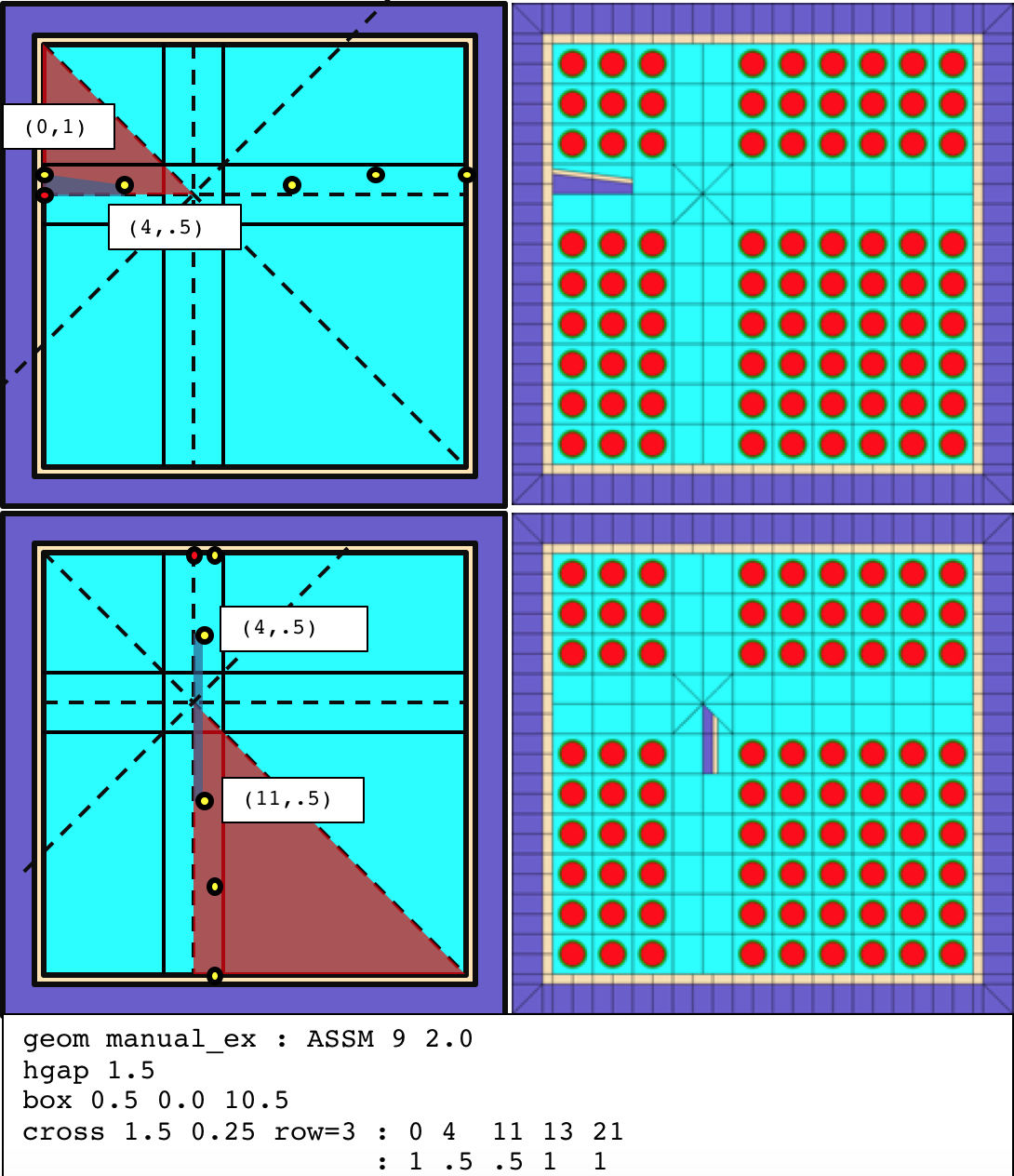
Fig. 3.2.6 Construction of the BWR cross geometry (full example shown later).
The cross structure is defined be a series of vertices (xi, yi). Shown as yellow points in the top left plot, the cross vertices are defined based on an origin displayed as the red point, which is the intersection of the inner west edge of the channel box and the horizontal line in that passes through the cross center.
The top plots demonstrate how Polaris inserts a section of the cross into the WNW octant. In the top left plot, the blue polygon is constructed based on the first two vertices defined on the cross card: (0.0,1.0) and (4.0,0.5). The intersection of the blue polygon and red polygon is inserted into the lattice and filled with cross interior material (Min). The liner is then inserted above the blue polygon, padded by the liner thickness (lthick), and clipped by WNW red polygon if needed.
Similarly, the bottom plots demonstrate insertion into the SSE octant. For SSE insertions, the origin and the cross vertices are rotated 90 degrees about the cross center. The blue polygon is constructed from the second and third vertices on the cross card: (4.0,0.5) and (11,0.5). The intersection of the blue polygon and red polygon is inserted into the lattice and filled with cross interior material (Min). The liner is then inserted above the blue polygon, padded by the liner thickness (lthick), and clipped by SSE red polygon if needed.
For each consecutive set of cross vertices, Polaris inserts a polygonal region into each of the 8 octants. The cross vertices are entered in the input as an x-values list followed by a y-values list of the same length. The coordinate system of the x- and y- lists is displayed in the top left plot of Fig. 3.2.6. The coordinate system is transformed based on the following rules for each octant:
WNW, ENE: no transform,
NNW, SSW: reflected across the diagonal line from NW to SE channel box corners,
NNE, SSE: rotated 90 degrees about the cross center, and
ESE, WSW: reflected across the line in the center of the horizontal cross gap.
The cross liner is inserted above the cross vertex values. The liner has a uniform thickness (lthick) and uniform material (Mcross). The uniform liner thickness is constructed with a miter joint at each cross vertex as shown in the following diagram:
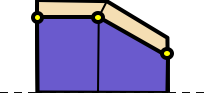
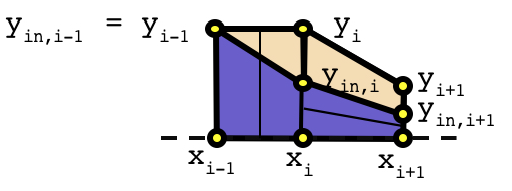
After the set of y-values on the cross card, an optional list of interior y-values can be specified. The length of the interior y-values list must be equal to the length of the x- and y- lists. The optional interior y-values list is used to split the polygons into two material regions as shown in the following diagram:
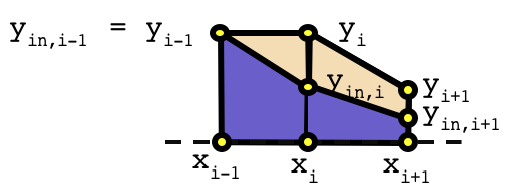
For the left-hand polygon, yin,i-1 is the same as yi-1, but yin,i is less than yi. In this scenario, the trapezoid region is filled with Min, and the triangular region is filled with Mcross. Similarly for the right-hand polygon, the lower trapezoid is filled with Min and the upper trapezoid is filled with Mcross. Note that the uniform liner above the y-values is not shown for simplicity.
The interior y-list values can be specified in one of two ways. First, a positive value may be entered that is greater than or equal to zero and less than or equal to the corresponding y-value. Second, a negative value may be entered that represents the relative distance of the interior y-value below the corresponding y-value. Note the Polaris input processor interprets “-0” different than “0”. “-0” implies that the internal y-value is the same as the y-value. “0” implies that the internal y-value is zero. If the interior y-list is omitted, the default for all internal y-values is “-0”, i.e., the polygon regions defined are completely filled with Min.
In addition to the interior y-values list, two additional lists can be used to refine the spatial mesh in the x- and y- directions. The list of nx- and ny- values subdivide the polygon regions along the x- and y- directions respectively. Both lists must have one less entry than the x-, y-, and yin- lists. If omitted, the default values for both the nx- and ny- lists are 1, i.e., no additional spatial refinement is applied to the polygon regions. For refinement in the y-direction, only the Min material is refined. The following diagram shows nx=2 refinement for the left polygon and ny=2 refinement for the right polygon:
The full cross example from Fig. 3.2.6 is displayed in the top left plot of Fig. 3.2.7. The bottom plot shows a centered cross structure with a diamond water box and empty pins surrounding the water box.
For additional examples, see the polaris.6.3 regression input files described at the beginning of Sect. 3.2.2.
See also:
box, system BWR, pinmap
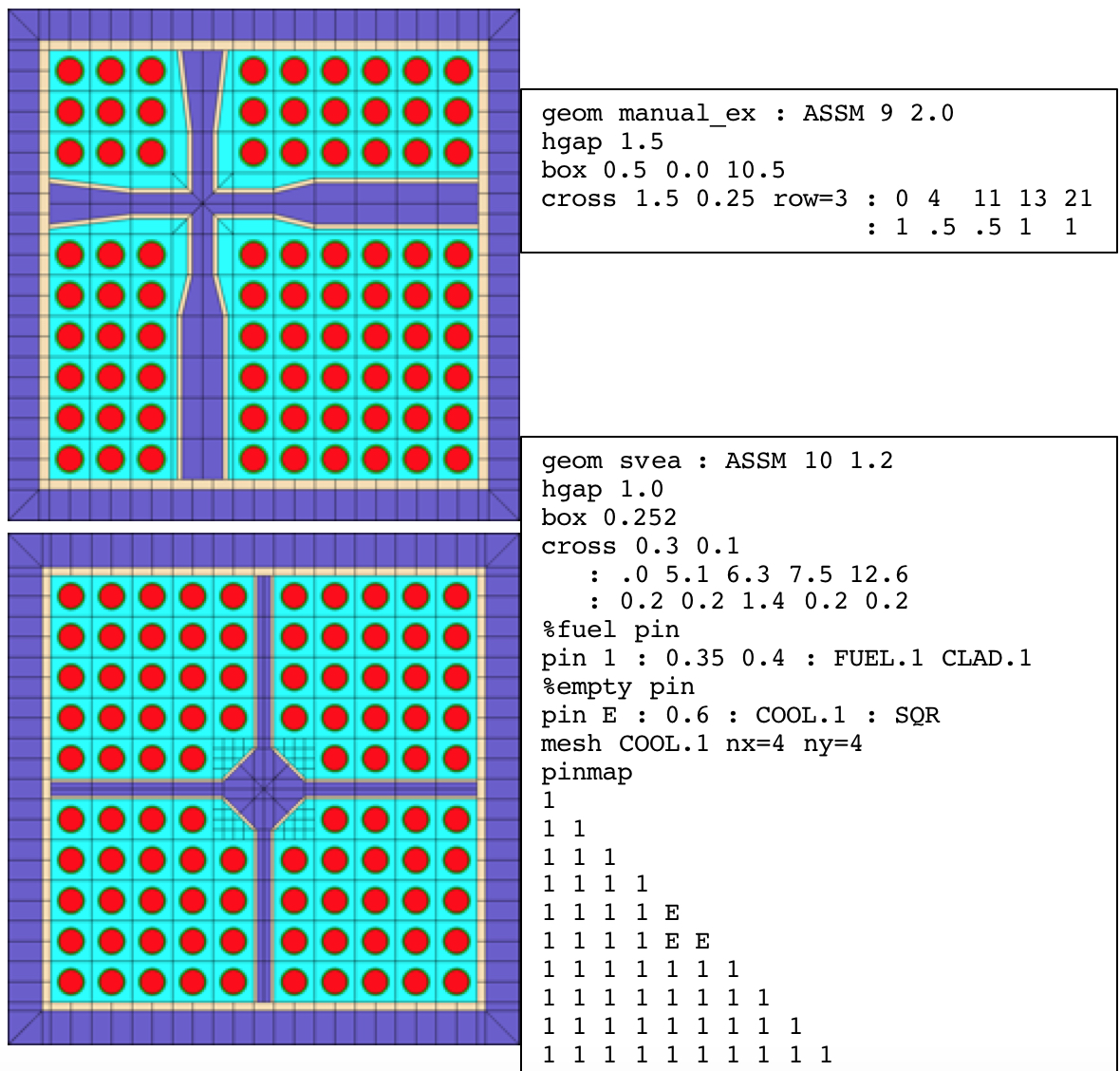
Fig. 3.2.7 Additional cross examples.
3.2.4.13. dxmap and dymap - pin-by-pin displacement maps
param |
type |
details |
default |
di |
Real |
pin center displacement value in the x or y direction (cm) |
0.0 |
Comments:
The dxmap and dymap cards displace pins from their natural position in the geometry (see comments for the pin card). If displacement maps are required, both the dxmap and dymap must be specified in the input and they must have the same length. However, the length of the displacement maps does not have to equal of length of the pinmap if the displacement maps have reduced symmetry. For integer-sized pins greater than 1, the displacement value should be entered in the northwest corner element of the size \(\times\) size subarray. For noninteger-sized pins, the displacement value should be in the corner element opposite of shared corner location. Note the following symmetry restrictions for the displacement maps:
dyi value must be zero on a horizontal symmetry line for an odd \(\times\) odd pinmap,
dxi value must be zero on a vertical symmetry line for an odd \(\times\) odd pinmap, and
dxi must equal dyi for an element on a diagonal symmetry line.
Examples:
dxmap
0.0 0.0 0.0 0.0 0.0
0.0 0.2 0.0 0.0 -0.2
0.0 0.0 0.0 0.0 0.0
0.0 0.0 0.0 0.0 0.0
0.0 0.0 0.0 0.0 0.0
dymap
0.0 0.0 0.0 0.0 0.0
0.0 0.0 0.0 0.0 0.0
0.0 0.0 0.0 0.0 0.0
0.0 0.2 0.0 0.0 0.0
0.0 0.0 0.0 -0.1 0.0

See also:
pinmap
3.2.4.14. mesh - advanced material dependent meshing options
param |
type |
name |
details |
default |
MSPEC |
MCLASS |MNAME |
material identifier |
||
nx |
Int |
# of x divisions |
must be >0 |
MeshNumX* |
ny |
Int |
# of y divisions |
must be >0 |
MeshNumY* |
nr |
Int |
# of radial rings |
must be >0 |
MeshNumRings* |
ns |
Int |
# of radial sectors |
must be nonzero |
MeshNumSectors* |
nf |
Int |
# of faces/pin |
must be >0 |
2** |
nd |
Int |
# of gap divisions |
must be >0 |
1 |
meshing multipliers |
||||
m |
Real |
global multiplier |
must be >0 |
1.0 |
mx |
Real |
x divisions |
must be >0 |
1.0 |
my |
Real |
y divisions |
must be >0 |
1.0 |
mr |
Real |
radial rings |
must be >0 |
1.0 |
ms |
Real |
radial sectors |
must be >0 |
1.0 |
mf |
Real |
faces/pin |
must be >0 |
1.0 |
md |
Real |
gap divisions |
must be >0 |
1.0 |
* The global mesh default values are set on the option <GEOM> card using the parameter name in the table above.
** Default number of faces per pin is 1. Default is 2 for system BWR or system PWR.
Examples:
mesh COOL : nr=3 ns=4 nx=2 ny=2 %coolant mesh: 3 ring, 4 sectors, 2 in x and y
mesh MOD.1 : nf=2 nd=4 %mesh used for wide gap (MOD.1): nf=2 nd=4
mesh MOD.2 : nf=2 nd=3 %mesh used for narrow gap (MOD.2): nf=2 nd=3
mesh FUEL : mr=2.0 %double the fuel radial mesh
mesh FUEL.2 : m=3.0 %triple all mesh values for FUEL.2
mesh CLAD : ms=0.5 %coarsen the clad sector mesh by a factor of 1/2.
Comments:
Polaris supports three different mesh types: 1) cylindrical mesh for CIR shapes in the pin card, 2) Cartesian mesh for SQR shapes in the pin card, and 3) a special Cartesian mesh for the region external to the pinmap region. As shown in the examples above, the mesh card is used to define, refine, or coarsen the mesh parameters for one or more of the mesh types associated with a given material class or material name. The default values for mesh parameters are defined through the option<GEOM> card and the system card. The default values on the option<GEOM> card are nr=1, ns=1, nx=1, ny=1, nf=1, nd=1, and MeshMult=1.0. The “MeshMult” multiplier from the option<GEOM> is a global mesh multiplier applied in conjunction with any material-specific multiplier (see option<GEOM> example for details). If system BWR or system PWR is applied, new default values include nf=2, ns=8, and nr=2 (only for the channel material class). If the final mesh value is noninteger, Polaris rounds down to determine the applied value.
See also: pin, system, option<GEOM>
3.2.4.15. detector - insert a detector geometry

|
Examples:
% declare detector materials
mat DET.2 : SS304 %transmitter
mat DET.3 : Al 2.7 %active material matrix (with u235 dopant not modeled)
% declare detector geometry
% - transmitting wire
% - active material
% - sheath
pin PD : 0.03 0.04 0.0787
: DET.2 DET.3 TUBE.1
%u235 for reaction rate material
mat DET.1 : detu 1e-5
comp detu : WT u235=99.9 u238=0.1
% place detector D1, described by pin PD
% into instrument tube pin.IT, at the center
% with signal proportional to fission in material DET.1
% but with flux inside material matrix DET.3
det D1 : pin.PD at=pin.IT loc=CENTER
: R(n,FIS) fmat=DET.3 rmat=DET.1
% enable detector output in XFile16
opt FG DetectorEdit=D1
Comments:
The detector card is not intended to produce arbitrary reaction rates, but model a simple, discrete detector inserted into the geometry. The first line of detector input defines the detector and where it should be placed. The second line of input defines the “signal” the detector produces. The most complex piece of the input is the reaction specificer (RSPEC) which denotes the units (reaction rate or energy rate), incident particle, and type of reaction. For example, neutron absorption rate would be denoted “R(n,ABS)” and gamma energy deposition rate “E(g,CAP)”.
The detector signal is collected for the material specified as the flux material, “fmat”. In some cases, e.g. PWR fission detector modeling, it is desirable to model a reaction rate for fissile material that does not exist, e.g. a U-235 fission rate in an gaseous fission chamber that does not have trace amounts of U-235 present in the composition definition. The optional reaction material, “rmat”, gives a way to specify this type of scenario. In this case the “fmat” would be a physical material in the model like the detector fill gas whereas the “rmat” would be another material (not present in the model) with composition and temperature defined appropriately.
3.2.5. Materials
A material contains two main types of information: (1) the composition, or distribution of nuclides, and (2) the properties which include basic (required) properties like density and temperature, and as well as (optional) properties like soluble poison content, void, or grid spacer smearing. The composition is defined by a composition card. The basic specification for a material is shown below.
[mat MNAME : CNAME [dens=*Real*] [temp=*Real*] [: properties]]
argument |
type |
name |
details |
default |
MNAME= MCLASS.MSUB |
Word. Int |
material name |
used to reference this material |
|
CNAME |
Word |
composition name |
||
dens |
Real |
density |
basic property units: g/cm3 |
composition reference density, if defined |
temp |
Real |
temperature |
basic property units: K |
293 |
properties |
Word=Value |
properties |
extra properties are defined with property cards |
no extra properties |
A material name has two parts, the material class, or MCLASS, and a member identifier, or MSUB. For example, FUEL.2 has an MCLASS=FUEL and an MSUB=2. All properties are defined by MCLASS. The composition referenced by CNAME is created with a composition card, as shown below.
[comp CNAME : CTYPE arguments]
argument |
type |
name |
details |
default |
CNAME |
Word |
composition name |
used to reference this composition later in materials and property definitions |
|
CTYPE |
- |
composition type |
||
General Composition Constructors |
||||
NUM |
number fraction |
|||
WT |
weight fraction |
|||
FORM |
Formula |
|||
CONC |
Concentrations |
|||
Reactor Composition Constructors |
||||
LW |
borated light water |
|||
UOX |
uranium oxide fuel |
|||
arguments |
- |
remaining arguments |
depends on CTYPE |
Additional properties are defined with the property card, which defines the property PNAME for a material class MCLASS. The property type, PTYPE, determines the remaining arguments.
prop PNAME MCLASS : PTYPE arguments
argument |
type |
name |
details |
default |
PNAME |
Word |
property name |
used to reference this property |
|
PTYPE |
PTYPE |
property type |
||
SOLP |
soluble poison |
used to define soluble boron content |
||
arguments |
- |
remaining arguments |
depends on PTYPE |
3.2.5.1. material - material initialization
argument |
type |
name |
details |
default |
MNAME |
Word. Int |
material name |
uses form MCLASS.MSUB |
|
CNAME |
Word |
composition name |
||
dens |
Real |
density |
basic property units: g/cm3 |
composition reference density |
temp |
Real |
temperature |
basic property units: K |
293 |
pi =vali |
PNAME=Value |
properties |
additional properties |
0 |
Examples:
% define a gas gap material
mat GAP.1 : FILLGAS
% define a 3.5% enriched fuel material
comp uox_e350 : UOX 3.5
mat FUEL.1 : uox_e350 dens=10.257 temp=900
% define a cladding material
mat CLAD.1 : ZIRC4
% define a guide tube material
mat TUBE.1 : SS304
% define a control rod material
mat CNTL.1 : AIC
Comments:
Material properties may be set on either a material card or on a state card. If a temperature is specified rather than a density, the “temp=” key must be used to skip over the density argument.
See also:
state, composition, property
3.2.5.2. composition<NUM|WT> – general atom/wt fraction

|
Examples:
% create a plutonium vector and then plutonium oxide
comp puvec : WT scale=PCT
Pu238=1.2
Pu239=63.3
Pu240=21.0
Pu241=8.6
Pu242=5.9
comp puox : FORM puvec=1 O=2
% create an 85/10/5 Ag/In/Cd composition
% using 10% In, 5% Cd,
% and filling the remainder up to 100% with Ag
comp aic : WT In=10 Cd=5 Ag=-100
Comments:
IDs in weight or number fraction-based compositions may be any of the following:
nuclide IDs (Int), e.g., 92235,
nuclide names (Word), e.g., U235 or u235,
element Z numbers (Int), e.g., 92,
element names (Word), e.g., U or u, or
other composition names (CNAME).
See also:
composition<FORM>
3.2.5.3. composition<FORM> - general chemical formula
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
FORM |
formula |
||
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
* |
structure |
Function |
structure |
see Structure names |
FREE |
idi =vali |
Word|Int=Real |
id/value pairs |
see acceptable id forms below values in atoms per molecule (e.g., H2O is given as “H”=2 “O”=1) |
*The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% define Gd2O3 using element names
% (using elements implies natural abundances used in isotopics)
comp gd2o3 : FORM Gd=2 O=3
% define Gd2O3 using 100% Gd 155
comp gd2o3 : FORM Gd155=2 O=3
% define D2O using nuclide IDs and element names
comp d2o : FORM 1002=2 8000=3
comp d2o : FORM H2=2 O=3
Comments:
IDs in formula-based composition may be any of the following:
nuclide IDs (Int), e.g., 92235,
nuclide names (Word), e.g., U235 or u235,
element Z numbers (Int), e.g., 92,
element names (Word), e.g., U or u, or
other composition names (CNAME).
See also:
composition<NUM|WT>, composition<CONC>
3.2.5.4. composition<CONC> - general number density
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
CONC |
concentration |
||
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
** |
idi =vali |
Int=Real |
id/value pairs |
see acceptable id forms below note: cannot use other CNAMEs for IDs in CONC input units: #/barn-cm |
**A reference density is automatically calculated from concentrations input. If specified, it will simply scale up concentrations linearly.
% pyrex composition
comp pyrex_e125 : CONC
5010=9.63266E-04
5011=3.90172E-03
8016=4.67761E-02
14028=1.81980E-02
14029=9.24474E-04
14030=6.10133E-04
% fuel composition
comp uox_e310_gd180 : CONC
92234=3.18096E-06
92235=3.90500E-04
92236=1.79300E-06
92238=2.10299E-02
64152=3.35960E-06
64154=3.66190E-05
64155=2.48606E-04
64156=3.43849E-04
64157=2.62884E-04
64158=4.17255E-04
64160=3.67198E-04
8016=4.53705E-02
% wet annular burnable absorber (WABA) composition
comp waba : CONC
5010=2.98553E-03
5011=1.21192E-02
6000=3.77001E-03
8016=5.85563E-02
13027=3.90223E-02
Comments:
IDs in concentration-based composition may be any of the following:
nuclide IDs (Int), e.g., 92235;
nuclide names (Word), e.g., U235 or u235;
element Z numbers (Int), e.g., 92;
element names (Word), e.g., U or u; and
SCALE-specific Nuclide IDs (Int), e.g., 3006000 (Only available for comp(CONC) card).
Other composition names (CNAME) cannot be used in a concentration definition. To easily ensure consistency of input when comparing codes, the composition input should be used in the concentrations described here. In all other cases, the other composition constructors are recommended because they are much simpler and easier to use.
See also:
composition<FORM>, composition<NUM|WT>
3.2.5.5. composition<LW> - borated light water
comp CNAME : LW [borppm=Real] [refdens=Real]
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
LW |
light water |
||
borppm |
Real |
boron |
parts per million by weight of natural boron (B) in light water (H2 O) |
* |
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
0.0 |
*The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% define a 600ppm boron moderator composition
comp mod_600ppm : LW 600
% same composition using FORM and WT
comp mod : FORM H=2 O=1
comp mod_600ppm : WT scale=PPM norm=yes
mod=1e6 B=600
Comments:
Internally, the borated light water composition is built from composition<FORM> and composition<WT> cards assuming natural boron. To use different boron isotopics such as depleted or enriched boron, the more general composition cards should be used.
See also:
composition<FORM>, composition<WT>
3.2.5.6. composition<UOX> -UO2 fuel
comp CNAME : UOX enr=Real [bu=Real] [refdens=Real]
param |
type |
name |
details |
default |
|---|---|---|---|---|
CNAME |
Word |
composition name |
||
CTYPE |
UOX |
uranium dioxide |
||
enr |
Real |
enrichment |
U-235 wt. % (see composition<ENRU> for formula) |
|
bu |
Real |
burnup |
only available for \(0\leq bu \leq 100\) units: GWd/MTU |
0* |
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
** |
*Note that the burnup parameter should not be specified. It is a future option to create a fixed, representative, burned fuel composition. The composition is interpolated using linear interpolation from an internal burnup- and enrichment-dependent data matrix.
**The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% define a 4.95% enriched fuel composition with a reference density
comp uox_495 : UOX 4.95 refdens=10.25
% same result as above
comp u_495 : ENRU 4.95
comp uox_495 : FORM u_495=1 O=2
Comments:
Internally, the UO2 composition is built from composition<FORM> and composition<ENRU> cards.
See also:
composition<ENRU>, composition<FORM>
composition<USI> – U2Si3 fuel
comp CNAME : USI enr=Real [bu=Real] [refdens=Real]
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
USI |
uranium silicide |
||
enr |
Real |
enrichment |
U-235 wt. % (see composition<ENRU> for formula) |
|
bu |
Real |
burnup |
only available for \(0\leq bu \leq 100\) units: GWd/MTU |
0* |
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
** |
*Note that the burnup parameter should not be specified. It is a future option to create a fixed, representative, burned fuel composition. The composition is interpolated using linear interpolation from an internal burnup- and enrichment-dependent data matrix.
**The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% define a 4.95% enriched fuel composition with a reference density
comp usi_495 : USI 4.95 refdens=11.54
% same result as above
comp u_495 : ENRU 4.95
comp usi_495 : FORM u_495=2 Si=3
Comments:
Internally, the U2Si3 composition is built from composition<FORM> and composition<ENRU> cards.
See also:
composition<ENRU>, composition<FORM>
3.2.5.7. composition<UN> - UN fuel
comp CNAME : UN enr=Real [n15enr=Real][bu=Real] [refdens=Real]
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
UN |
uranium nitride |
||
enr n15enr |
Real Real |
enrichment nitrogen enrichment |
U-235 wt. % (see composition<ENRU> for formula) N-15 wt. % |
100%* |
bu |
Real |
burnup |
only available for \(0\leq bu \leq 100\) units: GWd/MTU |
0** |
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
*** |
* Natural nitrogen is 0.4 wt. % 15N, but the reactivity penalty of 14N warrants using the highest 15N composition possible.
**Note that the burnup parameter should not be specified. It is a future option to create a fixed, representative, burned fuel composition. The composition is interpolated using linear interpolation from an internal burnup- and enrichment-dependent data matrix.
***The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% define a 4.95% enriched fuel composition with a reference density
comp usi_495 : UN 4.95 refdens=11.3
% same result as above
comp u_495 : ENRU 4.95
comp un_495 : FORM u_495=1 7015=1
% define a 3.25% enriched fuel composition with a reference density and
% specify natural 15N composition
Comp un_495 : UN 3.25 refdens=11.3 n15enr=0.4
Comments:
Internally, the uranium nitride composition is built from composition<FORM>, composition<WT>, and composition<ENRU> cards.
See also:
composition<ENRU>, composition<FORM>
3.2.5.8. composition<ENRU> – enriched uranium
comp CNAME : ENRU enr=Real [refdens=Real]
param |
type |
name |
details |
default |
CNAME |
Word |
composition name |
||
CTYPE |
UOX |
uranium dioxide |
||
enr |
Real |
enrichment |
*U-235 wt. % |
|
refdens |
Real |
reference density |
default density for materials using this composition units: g/cm3 |
** |
*The following formula from [POLARISGod14] is used to determine the 234U and 236U wt% from the 235U enrichment. Note that this formula is only valid for U-235 enrichments less than 10 wt%.wu234 = 0.007731*(enr) 1.0837wu236 = 0.0046*enrwu238 = 100 - wu234 - enr - wu236**The density property must be defined for each material either explicitly on the material card itself or implicitly through the “reference density” of the material’s composition.
Examples:
% 5% enriched metal fuel
comp umetal : ENRU 5
Comments:
This composition for enriched uranium is used internally to create uranium oxide, silicide, and nitride fuels using the composition<UOX>, composition<USI>, and composition<UN> cards.
See also:
composition<UOX>, composition<USI>, composition<UN>
3.2.5.9. composition library (pre-defined)
The Polaris composition library contains predefined compositions that may be used without a constructor by simply referencing the CNAME below. Each predefined library composition has a reference density, so it can be used directly on a material card.
CNAME |
Description |
|---|---|
H2O B4C Er2O3 Gd2O3 SiC ZrH Zr5H8 ZrH2 fillgas Cr2O3 Al2O3 BeO |
light water with structure=BOND(H2O) Boron carbide burnable poison material Erbium oxide burnable poison material Gadolinium oxide burnable poison material Silicon carbide zirconium hydride alloy with structure=CRYS(orthorhombic_zrh) zirconium hydride alloy with structure=CRYS(cubic_zrh) zirconium hydride alloy with structure=CRYS(tetragonal_zrh) Helium gas Chromium dioxide (chromia, Cr2O3) Aluminum dioxide (alumina, Al2O3) Beryllium dioxide (beryllia, BeO) |
CNAME |
Description |
|---|---|
aic pyrex zirc2 zirc4 ss304 ss316 inc718 water |
Ag-In-Cd control rod absorber material Pyrex glass Zircaloy-2 clad material Zircaloy-4 clad material Stainless Steel 304 Stainless Steel 316 Inconel 718 H2O with trace amount of boron |
pyroc |
Pyrolytic carbon, C with structure=CRYS(pyrolytic_c) |
graphite |
Graphite, C with structure=CRYS(hexagonal_c) |

|
Note
The cross section IDs can only be used on composition cards with the CONC variant to input number densities directly.
3.2.5.10. property<SOLP> - soluble poison by weight
prop PNAME M1 … : SOLP poison [scale=<PPM>|PCT|ABS]
param |
type |
name |
details |
default |
PNAME |
Word |
property name |
property value p \(\geq\) 0 |
|
M1 … |
MCLASS |
material class |
one or more material classes to gain this property |
|
PTYPE |
SOLP |
soluble poison |
||
poison |
CNAME |
soluble poison composition name |
||
scale |
PCT|ABS|PPM |
scaling factor |
all values are divided by this factor PCT: percentage (divide by 100) PPM: parts per million (divide by 1e6) ABS: absolute (divide by 1) |
PPM |
Examples:
% define a soluble boron property for moderator
% and coolant material classes
% using natural boron
prop boron MOD COOL : SOLP B
% investigate coolant crud/impurity activation
% 1. define a general impurity property to mix in coolant,
comp crud : NUM Ni=12.7 Cr=2.3 Fe=-100 %mostly Fe
prop impurity COOL : SOLP crud
% 2. create coolant material with 100ppm of crud
mat COOL.1 : LW dens=0.75 : impurity=100
% 3. make sure to "deplete" coolant so crud gets activated
deplete COOL=true
Comments:
None
See also:
pinmap, control, insert
3.2.5.11. property<DOPANT> - fuel dopant by weight
prop PNAME M1 … : DOPANT dopant [scale=<PPM>|PCT|ABS]
param |
type |
name |
details |
default |
PNAME |
Word |
property name |
property value \(p\geq 0\) |
|
M1 … |
MCLASS |
material class |
one or more material classes to gain this property |
|
PTYPE |
DOPANT |
fuel dopant |
||
dopant |
CNAME |
Fuel dopant composition name |
||
scale |
PCT|ABS|PPM |
scaling factor |
all values are divided by this factor PCT: percentage (divide by 100) PPM: parts per million (divide by 1e6) ABS: absolute (divide by 1) |
PPM |
Examples:
% define three dopants properties for the fuel
% using predefined Cr2O3, Al2O3, and BeO
% then dope (deferred definition) fuel with 0.3% chromia,
% 0.2% alumina, 0.1% beryllia
prop Cr2O3 FUEL: DOPANT Cr2O3
prop Al2O3 FUEL: DOPANT Al2O3
prop BeO FUEL: DOPANT BeO
mat FUEL.1 : uox_e310 temp=565 : Cr2O3=1000
% can also set the values through system properties
% works for both PWR and BWR
=polaris
title "pincell with UOX fuel doped with Cr2O3, Al2O3, and BeO"
lib "broad_lwr"
sys PWR
geom wec17 : ASSM 1 1.26
comp uox_e310 : UOX 3.10
mat FUEL.1 : uox_e310 10.5 : cr2o3=3000 al2o3=2000 beo=1000
pin 1 : 0.4096 0.418 0.475 : FUEL.1 GAP CLAD
end
Comments:
None
See also:
solp
3.2.5.12. property<TWOPHASE> - density property used to control two phase mixtures
property PNAME MCLASS : TWOPHASE liqden=Real vapden=Real
param |
type |
name |
details |
default |
PNAME |
String |
name for this property |
Name with which to refer to this TWOPHASE property in other cards. |
|
MCLASS |
MCLASS |
material class(es) for which this property is valid |
Generally COOL or MOD but can be any material. |
|
liqden vapden |
Real Real |
density of the liquid density of the vapor |
In g/cm3. In g/cm3. |
Examples:
% define a TWOPHASE combination for moderator and coolant
prop vf MOD COOL : TWOPHASE 0.7 0.04
Comments:
The two-phase property is used to model materials that exist in two phases, such as water existing in a liquid or gas. This is typically used for water in BWR systems where the density of the water changes as one moves axially through the core. This property is defined by the liquid and vapor density. Later, in the state or history cards, a number between 0% and 100% is entered and the final density is calculated by interpolating between the liquid and vapor density values. The only requirement is that the liquid density must be greater than the vapor density.
3.2.5.13. deplete - material depletion and decay
deplete M1=Bool M2=Bool … Mi=Bool … MN=Bool
param |
type |
name |
details |
default |
Mi |
MNAME|MCLASS |
list of material names or material classes |
use ALL for all materials |
Note
Only one deplete card is allowed in an input. ALL only applies in the first position.
Examples:
% turn on depletion/decay for two new materials
sys PWR
deplete MyMaterial=true MyOtherMaterial=true
% activate/deplete/decay every material
deplete ALL=true
% impose strict conditions
sys PWR
deplete ALL=false FUEL=true CLAD=true
Comments:
The deplete card not only instructs Polaris to deplete a material, but also to solve the Bateman equations with ORIGEN for that material. Thus if the flux/power is zero, only materials that are flagged to “deplete” will undergo decay. The deplete card modifies the depletables included in a system card to avoid the situation in which “deplete MyMaterial=true” would make only MyMaterial depletable. Thus to completely re-specify the depletable materials, “ALL=false” should be used as the first argument. This is in contrast to the basis card, which completely specifies a new power basis.
See also:
material, shield, basis
3.2.5.14. basis - power basis materials
basis M1=Bool M2=Bool … Mi=Bool … MN= Bool
param |
type |
name |
details |
default |
Mi |
MNAME|MCLASS |
list of material names or material classes |
use ALL for all materials |
ALL |
Note
Only one basis card is allowed per input. ALL is only allowed in the first position.
Examples:
% use only FUEL materials as the basis
basis ALL=no FUEL=YES
% Specify FUEL.3 as the basis
basis ALL=no FUEL.3=YES
Comments:
The basis card is used to specify the materials to use in power normalization. By default, the energy release from all materials is taken into account, including (n,gamma) reactions in structural materials such as cladding. It is not recommended to change the default of ALL in most situations. Exceptions include (1) when comparing results to other codes that only use fuel in the basis and (2) fixing the power in a specific pin is known-a material should be created only for that pin, and the power basis should be specified for that material only. The basis card overrides any power basis imposed by a system card. Thus it behaves differently than a deplete card, which is combined with depletable materials imposed by a system card.
See also:
material, shield, deplete
3.2.5.15. shield - cross section self-shielding expansion specification
shield M1=XTYPE M2=XTYPE … Mi=XTYPE … MN= XTYPE
param |
type |
name |
Details |
default |
Mi |
MNAME| MCLASS |
list of material names or material classes |
use ALL for all materials |
|
XTYPE |
N|P|R|S |
self-shield ing expansion type |
shield across various mesh elements N: no expansion P: pins R: rings (P implicit) |
R |
Note
Only one shield card is allowed per input. ALL is only allowed in the first position.
Examples:
%create a unique self-shielded FUEL cross sections in each pin
%consider all other materials to have a single self-shielded cross section
shield ALL=N FUEL=P
%assess effect of self-shielding each pin's cladding
shield CLAD=P
%re-specify self-shielding to be P by default, R for the FUEL
shield ALL=P FUEL=R
Comments:
The shield card controls how materials are internally expanded for self-shielding purposes. By default, Polaris expands all materials across pins and rings (R). For example, a fuel region defined on a pin card as having 10 rings will be expanded internally to have 10 different self-shielded cross sections. Because the R option also implicitly includes the P option, each instance of that pin will also get different cross sections.
When using specific systems (e.g., system PWR), this card is generally not needed. The shield card modifies the self-shielding options included in a system card. Thus, to completely re-specify the expansion, use “ALL=N” as the first argument. This is in contrast to the basis card, which completely specifies a new power basis.
See also:
material, deplete, system
3.2.6. State
The idea of a “state” or “statepoint” is a standard concept in lattice physics calculations. In Polaris, the concept of state is mostly tied to the values of material properties. The base state for a calculation is determined as follows:
The base state is initialized with any property values set on material cards.
The base state is updated with any state cards that apply to ALL.
The base state is updated with any other state cards, and the power card is used to set the base state power.
This sequence ensures that the state does not change, even if the order of inputs changes. A time or burnup card is then used to initiate a calculation as a function of time or burnup, thus producing a sequence of states. A branch block is used to perform branches off the base state at specific times or burnups.
single value mode:
array mode with M burnup/time values, only in history block:
param |
type |
name |
details |
default |
NAME |
MNAME|MCLASS, or INAME, or GNAME |
material name/class insert/cont rol geometry |
use ALL for all materials in=yes|no pres=yes|no |
|
pi |
PNAME |
property name |
||
vali |
Value |
property value |
Examples:
state ALL : temp=293
COOL : dens=1.0 boron=1100
BPMAP : in=yes
read history
%--------------------------------------%
% cycle 1
%--------------------------------------%
pow 39
state ALL : temp=600
COOL : boron=900 850 700 600 400
dens =0.6
FUEL : temp =900 910 920 890 880
dt 50 50 100 100 200
pow 0
dt 70
%--------------------------------------%
% cycle 2
%--------------------------------------%
state BPMAP : in= no
COOL : boron=900 850 700 600 300
FUEL : temp =900 910 920 890 880
pow 42 41 38 39 37
dt 50 50 50 100 250
pow 0
dt 80
end history
Comments:
The state card is used to specify properties for different materials, control maps, control blades, and insert maps. The state card can specify one or multiple properties simultaneously. The property specifications can either be a single value or multiple values, each value corresponding to a burnup/time step in the burnup/time input card (bu|bui|dbu|t|ti|dt).
The state card can be used outside a history block or inside a history block. Outside the history block, i.e., at the “root” input level, a single state card can initialize property values to a single value. If one or more power histories are defined at the root input level, the state properties are constant throughout the calculation. The state properties are only modified for branch calculations.
Time-dependent state properties are allowed through the history block. One or more power histories are allowed inside a history block. Each power history contains a power card (single value or array value) and a burnup/time card (bu|bui|dbu|t|ti|dt). Before the burnup/time card, a state card can be used to define one or more state properties. The property specifications are either a single value or an array of values that correspond to each burnup/time step. If a property is omitted in a state card, the value is defined based on the following precedence:
the last value specified through the closest preceding state card in a given history block, or
the value specified in the state card at the root input level, or
the default property value.
By default, all control or insert geometries are not inserted (in=no). Material property defaults are defined on the material card or through the system card.
See also:
history, system
3.2.6.1. power - total power
pow [: p1 p2 … pi … pN]
param |
type |
name |
details |
default |
pi |
Real |
list of specific powers in W/g initial heavy metal |
0 |
Examples:
% set power to 35 W/gIHM
power 35.0
% provide a power history
% must have same number of values as following time/burnup card
power : 35.0 40.0 45.0 45.0 40.0 5.0 0.0
time : 10 20 30 40 50 60 70
Comments:
The power card specifies the total power of the basis materials specified by a basis card. The power value may be specified only once.
See also:
t, bu, history, basis, state<MNAME>
3.2.6.2. bu - initiate calculation with cumulative burnups
param |
type |
name |
default |
bi |
Real |
list of burnups |
0 |
units |
GWD/MTIHM| MWD/MTIHM |
burnup units |
GWD/MTIHM |
Examples:
% simple depletion case with constant power and absolute/cumulative burnups
power 40
bu 5 10 15 20 30 40 50 60 80
% using MWd/MTIHM units with variable power
% 40 W/gIHM for 0→5000 MWD/MTIHM, then 30 W/gIHM for 5000→10000 MWD/MTIHM
power 40 30
bu 5000 10000 MWD/MTIHM
% combine burnup and time cards
% 20 W/gIHM for 0→5 then 5→10 GWD/MTIHM steps, then
% 40 W/gIHM for a 5-day step then 30 W/gIHM for a 5-day step
power 20
bu 5 10 GWD/MTIHM
power 40 30
dt 5 5 DAYS
Comments:
The bu card initiates a calculation for a given sequence of cumulative/absolute burnups. A burnup or time card usually follows a power card, the two effectively specifying the power history. If multiple burnups are given, then the power card must have either a single power or a list of powers the same size as the list times. A value of 0 is implicit at the beginning of the first burnup list. Multiple burnup/time cards may be specified in an input. This can be convenient for switching units or changing from burnup-based to time-based depletion. Internal automatic substeps are always in effect unless modified with the option<DEPL> card.
See also:
t, dt, ti, bui, dbu, power, option<DEPL>, branch, deplete
3.2.6.3. bui - initiate calculation with cumulative burnups (with restart)
param |
type |
name |
default |
bi |
Real |
list of burnups |
0 |
units |
GWD/MTIHM| MWD/MTIHM |
burnup units |
GWD/MTIHM |
Examples:
power 30
bui 5 10 %equivalent to: bu 5 10
power 40
bui 5 10 %equivalent to: bu 15 20
Comments:
The bui card initiates a calculation for a given sequence of cumulative burnups. If only one burnup list is provided, the bui card is identical to the bu card. For any subsequent burnup list, the bui card specifies cumulative burnups that restart at zero at the beginning of each list (see example above).
See also:
t, dt, ti, bu, dbu, power, option<DEPL>, branch, deplete
3.2.6.4. dbu - initiate calculation with incremental burnups
param |
type |
name |
details |
default |
bi |
Real |
list of incremental burnups |
0 |
|
units |
GWD/MTIHM| MWD/MTIHM |
burnup units |
GWD/MTIHM |
Examples:
% incremental burnups equivalent to
% power 40
% bu 0 5 10 15 20 30 40 50 60 80
power 40
dbu 5 5 5 5 10 10 10 10 20
Comments:
The dbu card initiates a calculation for a given sequence of incremental burnups. Otherwise, it is identical to the bu card for specifying cumulative burnups.
See also:
t, dt, ti, bu, bui, power, option<DEPL>, branch, deplete
3.2.6.5. t - initiate calculation by cumulative time
param |
type |
name |
details |
default |
ti |
Real |
list of times |
0 |
0 |
units |
SECONDS | MINUTES | HOURS | DAYS | YEARS |
time units |
DAYS |
Examples:
% burn with 40 W/gIHM for 300 days in 100-day increments
power 40
t 100 200 300
% simulate 2 cycles of time-dependent irradiation with shutdown cooling
% note that time defaults to DAYs
%
% cycle 1
power 40 30 30 30
t 100 200 300 400
power 0
t 415
%
% cycle 2
power 30 20 20 20
t 515 615 715 815
power 0
t 830
Comments:
The t card initiates a calculation for a given sequence of cumulative/absolute times. One of the time cards (t, dt, or ti) is required to model periods of decay in conjunction with power 0. Otherwise, the time card t is similar in functionality to the burnup bu card but with different units.
See also:
ti, dt, bu, bui, dbu, power, option<DEPL>, branch, deplete
3.2.6.6. ti - initiate calculation by cumulative time (with restart)
param |
type |
name |
details |
default |
ti |
Real |
list of times |
0 |
0 |
units |
SECONDS|MINUTES| HOURS|DAYS |YEARS |
time units |
DAYS |
Example with t card:
% cycle 1
power 40
t 100 200 300 400
power 0
t 415
% cycle 2
power 30
t 515 615 715 815
power 0
t 830
Equivalent example with ti card:
% cycle 1
power 40
ti 100 200 300 400
power 0
ti 15
% cycle 2
power 30
ti 100 100 100 100
power 0
ti 15
Comments:
The ti card initiates a calculation for a given sequence of cumulative times. If only one time list is provided, the ti card is identical to the t card. For any subsequent time list, the ti card specifies cumulative times that restart at zero at the beginning of each list (see example above).
See also:
t, dt, bu, bui, dbu, power, option<DEPL>, branch, deplete
3.2.6.6.1. dt - initiate calculation by incremental time
param |
type |
name |
details |
default |
ti |
Real |
list of times |
0 |
|
units |
SECONDS|MINUTES| HOURS|DAYS|YEARS |
time units |
DAYS |
Examples:
% burn with 40 W/gIHM for 300 days in 100-day increments equivalent to
% power 40
% t 100 200 300
power 40
dt 100 100 100
% decay for 30 minutes
power 0
dt 30 MINUTES
Comments:
The dt card is identical to the cumulative time card t except that the values given are incremental.
See also:
t, ti, bu, bui, dbu, power, option<DEPL>, branch, deplete
3.2.6.7. branch - instantaneous change
param |
type |
name |
details |
default |
BNAME |
Word |
branch name |
DEFAULT |
|
allowed cards in branch block |
||||
add |
- |
- |
adds a list of states to branch on |
Examples:
% fuel temperature and boron branches (results in 7 total states)
read branch
add FUEL : temp=800 1000 1200
add COOL : boron=0 400 800 1400
end branch
% branch to different
% fuel temp/coolant temp/coolant density, synchronizing
% states (results in 3 total states)
read branch
% state 1 2 3
add FUEL : temp=800 1000 1200
COOL : temp=565 585 620
COOL : dens=0.73 0.71 0.68
end branch
Comments:
The branch card initiates so-called “branch” calculations, i.e., instantaneous changes of state at specific burnups/times during the base depletion sequence of calculations. The syntax for the add card is identical to the state card except, instead of taking a list of different properties and their values, it takes a single property and a list of values. Note that a time or burnup card is not necessary-if not found, branches will be performed at every burnup/time specified in the base state. The initial state for any branch card is the base state as specified in the main file. This means branch cards have no knowledge of one another.
See also:
add, bu, t, title
3.2.6.8. history - time-dependent history
param |
type |
name |
details |
default |
HNAME |
Word |
history name |
DEFAULT |
|
allowed cards in history block |
||||
power |
- |
- |
specific power |
|
state |
- |
- |
state properties |
|
bu|bui|dbu|t|ti|dt |
- |
- |
burnup or time |
Examples:
state ALL : temp=293
COOL : dens=1.0 boron=1100
BPMAP : in=yes
read history
%--------------------------------------%
% cycle 1
%--------------------------------------%
pow 39
state ALL : temp=600
COOL : boron=900 850 700 600 400
dens =0.6
FUEL : temp =900 910 920 890 880
dt 50 50 100 100 200
pow 0
dt 70
%--------------------------------------%
% cycle 2
%--------------------------------------%
state BPMAP : in= no
COOL : boron=900 850 700 600 300
FUEL : temp =900 910 920 890 880
pow 42 41 38 39 37
dt 50 50 50 100 250
pow 0
dt 80
end history
Comments:
The history card initiates a time-dependent calculation with user defined power history and time-dependent material or geometry properties. Each history block is independent from one another. Each history calculation generates an ORIGEN binary concentration file with the name filename_hname.f71 where filename is the root name of the input file and hname is the name for the history. Similarly, if few-group cross section files are requested, the filenames are filename.hname.t16. If ORIGEN binary library files are requested, the names are filename.hname.XXX.f33 where XXX is the name of the material (or system) for the corresponding library.
History calculations are also allowed in conjunction with branch calculation. At this time, all history calculations will perform all branch calculations defined in the input file. Selection of the burnup values for branch calculations is available on the option <FG> card.
See also:
state, branch, option<FG>, power, bu, bui, dbu, t, ti, dt
3.2.6.9. add<MNAME> - material branch
param |
type |
name |
details |
default |
MNAME|MCLASS |
- |
material name or material class |
use ALL for all materials |
|
incr |
Bool |
increment |
values are added to reference value |
false |
scale |
ABS|PCT |
Scaling |
scaling ABS: absolute units PCT: percentage units |
ABS |
PNAME |
- |
property name |
||
vali |
Value |
list of property values |
Examples:
% fuel temperature branches using incremental
% changes from the base state of 900 K
state FUEL : temp=900
read branch
add FUEL incr=true : temp=-200 -100 +100 +200 +500
end branch
% material properties may be varied together (synchronized)
% by chaining additional material/properties together
% the first block below results in 2 states
% the second is 6 states
read branch
add FUEL : temp=900 1200
FUEL : dens=10.4 10.3
COOL : dens=0.7 0.65
end branch
read branch
add FUEL : temp=900 1200
add FUEL : dens=10.4 10.3
add COOL : dens=0.7 0.65
end branch
Comments:
The add card is only valid inside a branch block. This version adds a set of branches for a specific material name (MNAME) or class (MCLASS). Branches are always with respect to the base state. Although similar to the state card, the add card has a single property name and a list of values. The state card has a list of property=value pairs.
See also:
material, state<MNAME>
3.2.6.10. add<INAME> - insert/control branch
add INAME : in=Bool1[Bool2]
param |
type |
name |
details |
default |
INAME |
- |
insert name or control element name |
||
in |
Bool |
list of insertion states |
“in=” is required |
Examples:
% branch to remove WABA inserts
state InsWABA4 : in=true
read branch
add InsWABA4 : in=false
end branch
% synchronize rods in with material branches (5 states)
read branch
add BankD : in=true false false false true
FUEL : temp=600 900 1200 2000 2000
end branch
% swap control banks
read branch
add BankB : in=true false false
BankC : in=false true false
BankD : in=false false true
end branch
Comments:
This form of the add card is required to add branches to insert/remove control elements or inserts. Given that only two possible states exist, specifying “true false” will result in a calculation at the other state not specified by the base state.
See also:
insert, control, state<INAME>
3.2.6.11. add<GNAME> - geometry branch
add GNAME : pres=Bool1[Bool2]
param |
type |
name |
details |
default |
GNAME |
- |
geometry name |
||
pres |
Bool |
list of geometry states |
“pres=” is required |
Examples:
% perform a reflector calculation on a branch
state ReflectorNode : pres=no
read branch
add ReflectorNode : pres=yes
end branch
Comments:
This form of the add card is required to add branches for new geometry, such as reflector calculations. Given that only two possible states exist, specifying “true false” will result in a calculation at the other state not specified by the base state.
See also:
geometry, state<GNAME>
3.2.7. Options
An extensive set of options is provided for manipulating the solvers and output. Most option cards support a key=value style of input, with reasonable defaults in place for all parameters.
3.2.7.1. option<KEFF> - eigenvalue
opt KEFF [key1=val1 key2=val2 … keyi=vali … keyN=valN]

|
Examples:
% change the MOC ray spacing for the
% eigenvalue calculation to 0.01 cm
opt KEFF RaySpacing=0.01
%P3 scattering
opt KEFF PnOrder=3
3.2.7.2. option<ESSM> - embedded self-shielding

|
param |
type |
details |
default |
|---|---|---|---|
MGi |
String |
Material group name for ESSM group i. All materials in the model with a material ID which contains the material group name will have their ESSM escape cross-section calculations performed together. The method used to performed these calculations will be designated by meti |
ALL |
meti |
String |
The ESSM calculation method to be used by material group MGi. Acceptable values are either “G” for full “Group-wise” treatment, or “I” for the Enhanced Neutron Current based tabular “Interpolation” approximation method. |
G |
Ei |
Real |
Energy values in units of eV which specify the energies to be used for tabular Interpolation. These values must be provided in ascending order. |
0.1 1 10 |
Examples:
% change within group solver to use source iterations
opt ESSM WithinGroupSolver=SOURCE
% change all CLAD to use the interpolation
% self-shielding method (faster)
opt ESSM RaySpacing=0.08
NumAzim=1
NumPolar=1
MinEnergy=1e3
MaxEnergy=1e7
: CLAD=I FUEL=G
: 10 100 1000 10000
3.2.7.3. option<BOND> - Bondarenko search
opt BOND [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
|---|---|---|---|
MaxIterations |
Int |
number of Bondarenko iterations (0 is disabled) |
0* |
ConvergenceXS |
String |
cross section used in Bondarenko convergence iterations “SIGA”: absorption “SIGT”: total |
“SIGA”** |
developer options (generally should not change) |
|||
IterationCriteria |
Real |
1e-3 |
|
TempSearchMaxIterations |
Int |
20 |
|
TempSearchCriteria |
Real |
1e-8 |
|
TempSearchEqual Tolerance |
Real |
1e-3 |
|
TempSearchMaxPower |
Real |
35.0 |
|
Sig0SearchMaxIterations |
Int |
20 |
|
Sig0SearchCriteria |
Real |
1e-8 |
|
Sig0SearchEqual Tolerance |
Real |
1e-3 |
|
Sig0SearchMaxPower |
Real |
35.0 |
|
* By setting the Bondarenko iterations >0, resonance interference effects may be taken into account. The default MaxIterations=0 effectively disables the Bondarenko resonance interference model.
**In SCALE, the transport cross section (“SIGT”) has historically been used in Bondarenko iterations.
Examples:
% introduce Bondarenko iterations on total cross section
opt BOND
MaxIterations=10
ConvergenceXS="SIGT"
3.2.7.4. option<DEPL> - depletion
opt DEPL [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type. |
details |
default |
|---|---|---|---|
TrackingSet |
String |
set of nuclides tracked in depletion calculations “None”: set of nuclides present in user input “Complete”: complete set of all nuclides available on ORIGEN data libraries |
“Complete” |
Solver |
String |
“MATREX”: legacy ORIGEN solution method “CRAM”: Chebyshev Rational Approximation Method |
“MATREX” |
Method |
String |
“PREDICTOR” “PREDICTOR_CORR ECTOR” |
“PREDICTOR_CORR ECTOR” |
StepRefinement |
Int |
divide the user input steps by this factor>0, i.e., refinement of 2 divides all steps by 2 (NOT ENABLED) |
1 |
NumSubsteps |
Int |
Number of internal substeps for depletion calculations |
4 |
DepleteMode |
String |
“BOSS” or “MOSS.” The depletion power renormalization is done at the beginning of each substep (BOSS) or the middle of each substep (MOSS) |
“BOSS” |
ArchiveF71 |
ALL|NONE |
Archive all of materials on the .f71 file, or do not archive any isotope inventories |
ALL |
ArchiveF33 |
ALL |NONE|SYSTEM |
Archive all of materials on the .f33 file, or no materials, or just the system average. |
SYSTEM |
Examples:
% Set the number of origen substeps per time steps to 2
% (may be useful for convergence studies)
opt DEPL
NumSubsteps=2
% disable the addition of depletion nuclides to input materials
opt DEPL TrackingSet="NONE"
% use CRAM solver
opt DEPL Solver="CRAM"
3.2.7.5. option<CRITSPEC> - critical spectrum
opt CRITSPEC [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
|---|---|---|---|
Mode |
String |
critical spectrum mode “SEARCH”: search for critical mode (k-eff=1) “SPECIFIED”: provide B2 below “NONE”: do not use critical spectrum |
“SEARCH” |
B2 |
Real |
value of critical buckling if Mode=”SPECIFIED “ units: cm-2 |
0.0 |
Method |
String |
critical spectrum system “B1”: solve the B1 equations “P1”: solve the P1 equations |
“P1” |
Examples:
% enable critical buckling search using B1 equations for a buckling of 1e-3
opt CRITSPEC
Mode="SPECIFIED"
B2=1e-3
Method="B1"
3.2.7.6. option<PRINT> - printing
opt PRINT [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
|---|---|---|---|
XSSummary |
Bool |
print a cross section summary in the output file |
yes |
CritSpecSummary |
String |
print critical spectrum summary “NONE”: no print out “BUCKLING”: limited buckling info “SPECTRUM”: full spectrum |
“BUCKLING” |
XFile16 |
Bool |
output a TRITON xfile016 nodal data library |
no |
InputDataContainer InputPropertySummary |
Bool Bool |
print out the input data container print out the user defined properties |
yes yes |
InputCompositionSummary |
Bool |
print compostion card input summary |
yes |
InputMaterialSummary |
Bool |
print material card input summary |
yes |
LibrarySummary |
Bool |
print cross section library summary |
no |
MaterialSummary |
Bool |
print material summary at each statepoint |
no |
FgNodalSummary |
Bool |
print few-group nodal output summary |
yes |
DeplF71Summary |
Bool |
print .f71 archive summary |
yes |
DeplF33Summary |
Bool |
print .f33 archive summary |
yes |
Examples:
% print the xfile016
% if input file is polaris.inp, file name will be polaris.x16
opt PRINT XFile16=yes
% print summaries
opt PRINT XSSummary=yes
CritSpecSummary="SPECTRUM"
InputCompositionSummary=yes
InputMaterialSummary=yes
LibrarySummary=yes
MaterialSummary=yes % disabled for now
3.2.7.7. option<FG> - few-group cross section generation
param |
type |
details |
default |
|---|---|---|---|
AdjointMode |
String |
type of adjoint calculation to use in few-group data generation “INFMED”: infinite medium adjoint “CRITICAL”: critical spectrum adjoint “UNIFORM”: uniform adjoint |
“INFMED” |
InvVelMode |
String |
weighting option for few-group inverse velocities “FORWARD”: forward flux weighting “ADJOINT”: adjoint flux weighting |
“FORWARD” |
DetectorEdit |
DNAME |
Name of the detector to use in XFile16 detector edits (see detector card) |
none |
bi |
Real |
list of burnups to include in output few-group cross section database, e.g., XFile16 output units: GWd/MTHM |
all burnups available |
Ei |
Real |
note descending order and only N-1 divisions are needed for an N group structure E0 is maximum energy (typically 2e7 eV) EN is minimum (typically 1e-5 eV) units: eV |
0.625 eV division (two groups) |
Examples:
% enable a detector edit to the XFile16 based on detector D1
opt FG : DetectorEdit=D1
% enable the critical spectrum adjoint
opt FG AdjointMode="CRITICAL"
%only include 0,10,15,20 GWd/MTHM burnups in few-group outputs, including XFile16
opt FG : 0 10 15 20
%redefine group energy divisions for 3 groups with divisions at 10 and 0.625 eV
opt FG : : 10 0.625
3.2.7.8. option<RUN> - run time
opt RUN [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
CheckOnly |
Bool |
check input and terminate |
true |
HomogenizeGrains |
Bool |
homogenize grains (DISABLED) |
false |
Examples:
% check input
opt RUN CheckOnly=true
3.2.7.9. option<GEOM> - geometry options
opt GEOM [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
|---|---|---|---|
NumPlotRays |
Int |
Number of rays used to generate the geometry PNG file |
1500 |
MeshMult |
Real |
Global mesh multiplier |
1.0 |
MeshNumRings |
Int |
Default number of radial rings for circular pin zones |
1 |
MeshNumSectors |
Int |
Default number of radial sectors for circular pin zones |
1 |
MeshNumX |
Int |
Default number of Cartesian x subdivisions for square pin zone |
1 |
MeshNumY |
Int |
Default number of Cartesian y subdivisions for square pin zone |
1 |
LegacyChannelMe sh |
Bool |
Use legacy radial mesh approach in the pin outermost circular zone |
false |
AreaTol |
Real |
1e-6 |
|
RelTol |
Real |
5e-8 |
|
AbsThresh |
Real |
5e-10 |
|
PlaneHitTol |
Real |
1e-13 |
|
PlaneParallelTol |
Real |
1e-13 |
|
PolyAcceptTol |
Real |
5e-10 |
|
PolyHitTol |
Real |
1e-9 |
|
PolyPointTol |
Real |
1e-12 |
|
UnitPointTol |
Real |
0.0 |
|
UnitLookAheadPointTol |
Real |
1e-9 |
Examples:
% Make the png file smaller
opt GEOM NumPlotRays=1000
% Double the mesh everywhere
opt GEOM MeshMult=2.0
% Default the ring mesh to 3
opt Geom NumMeshRings=3
See also: mesh
3.2.7.10. option<GAMMA> - gamma transport calculation
opt GAMMA [key1=val1 key2=val2 … keyi=vali … keyN=valN]

|
Examples:
% change the solver method to source iteration
opt GAMMA
Solver="SOURCE"
3.2.7.11. option<DATA> – data libraries
opt DATA [key1=val1 key2=val2 … keyi=vali … keyN=valN]
key |
value type |
details |
default |
|---|---|---|---|
YieldResource |
String |
Fission yield data |
yield.data |
ReducedYieldResource |
String |
Reduced fission yield data |
lwr_yields.h5 |
DecayResource |
String |
ORIGEN decay data |
decay.data |
NuclideResource |
String |
Nuclide mass and abundance data |
nuclide.data |
KineticsResource |
String |
Kinetics data |
kinetics.h5 |
ReactionResource |
String |
JEFF reaction data for ORIGEN |
reaction.data |
TransitionDefinition |
String |
Master ORIGEN transition data |
transition.def |
Examples:
% change the data resources to different values
% Can be aliases from FileNameAliases.txt or filenames
opt DATA
KineticsResource="kinetics_Tuttle.h5"
ReactionResource="n302.reaction.data"
3.2.8. System
The system cards provide a way to initialize a set of defaults to simplify input and add robustness for a well-known and well-characterized system.
system STYPE
argument |
type |
name |
STYPE |
system type |
|
PWR |
pressurized water reactor |
|
BWR |
boiling water reactor |
The system card performs the following actions:
defines a set of materials and properties, imposing standard names for the materials and properties;
warns user of potential mistakes; and
uses heuristics to modify unspecified mesh and solver options for robust results.
3.2.8.1. system<PWR> - pressurized water reactor
sys PWR
Definitions

|
Description
Twelve reactor materials are initialized with compositions and densities from the predefined composition set. In most cases, all that remains is to define fuel materials, all material temperatures, and properties such as COOL/MOD soluble boron and density. Note that some rules are based on naming conventions. For example, burnable poisons (the material class BP) are declared to be depletable materials, whereas the CNTL (control elements) class of materials is not.
Examples:
% a complete input file for a PWR pincell model
=polaris
system PWR
geom MyPin : ASSM 1 1.5
comp f35 : UOX 3.5
mat FUEL.1 : f35 dens=10.25
pin 1 : 0.5 0.6 : FUEL CLAD
state ALL : temp=565
state MOD : dens=0.743
state COOL: dens=0.743
state ALL : boron=600
power 40
burn 0 0.1 0.2 0.5 1 2 5 10 15
20 25 30 35 40 45 50 55 60
end

3.2.9. Sample Problems
Within the SCALE distribution, 31 Polaris sample problems are provided
to demonstrate the differences in calculation and geometry options, and
21 sample problems consider the Consortium for Advanced Simulation of
Light Water Reactors (CASL) Virtual Environment for Reactor Applications
(VERA) benchmark problems for pin cell and lattice configurations
described in [POLARISRE13]. The VERA pin cell problems are identified as
polaris_1a.inp through polaris_1e.inp. The VERA lattice
problems are identified as polaris_2a.inp through polaris_2p.inp.
The remaining ten sample problems are described as follows:
polaris_TMI1_Cycle1-2.inp – 15 \(\times\) 15 PWR geometry model with branch block definition for lattice physics calculations
polaris_bench_taka3_sf97-4_assm.inp, polaris_bench_taka3_sf97-4_pin.inp – Takahama UOX depletion benchmark for radiochemical assay NT3G24-SF97-4 described in [POLARISBS96].
polaris.6.3_rca_pwr.inp – Additional Takahama UOX depletion benchmark with time-dependent boron concentraion defined in a history block.
polaris_bwr10x10.inp, polaris_bwr7×7.inp – example BWR geometry models for 10 × 10 and 7 x 7 fuel.
polaris_dv1a.inp – simple PWR pin cell depletion calculation.
polaris_twophase.inp – simple BWR 8 x 8 fuel example to demonstrate the TWOPHASE property.
polaris_dopants.inp – pin cell demonstrations of built-in dopant properties for LWR fuel.
polarisHatchT1ASimple.inp - BWR model with detector model in narrow-narrow corner.
References
- POLARISBS96
S. M. Bowman and T. Suto. Scale-4 analysis of pressurized water reactor critical configurations: Volume 5, North Anna Unit 1 Cycle 5. Technical Report, Oak Ridge National Laboratory, Oak Ridge, TN (USA), 1996.
- POLARISGod14
Andrew T. Godfrey. VERA core physics benchmark progression problem specifications. Consortium for Advanced Simulation of LWRs, 2014.
- POLARISJWM+21
Matthew A. Jessee, William A. Wieselquist, Ugur Mertyurek, Kang Seog Kim, Thomas M. Evans, Steven P. Hamilton, and Cole Gentry. Lattice physics calculations using the embedded self-shielding method in polaris, part i: methods and implementation. Annals of Nuclear Energy, 150:107830, 2021.
- POLARISRE13
B. Roque and M. Erlund. International Comparison of a Depletion Calculation Benchmark on Fuel Cycle Issues, Results from Phase 1 on UOx Fuels. Technical Report, NEA/NSC/DOC, 2013.
- POLARISWK12
Mark L. Williams and Kang Seog Kim. The embedded self-shielding method. In PHYSOR 2012. 2012.
3.2.10. Appendices
- 3.2.10.1. SCALE 6.2 Polaris Input Format
- 3.2.10.1.1. box – channel box
- 3.2.10.1.2. pin – pin or pincell
- 3.2.10.1.3. bu – initiate calculation with cumulative burnups
- 3.2.10.1.4. dbu – initiate calculation with incremental burnups
- 3.2.10.1.5. t – initiate calculation by cumulative time
- 3.2.10.1.6. dt – initiate calculation by incremental time
- 3.2.10.1.7. option<ESSM> - embedded self-shielding
- 3.2.10.1.8. option<FG> – few-group cross section generation
- 3.2.10.1.9. state<MNAME> – material state
- 3.2.10.1.10. state<INAME> – insert/control state
- 3.2.10.1.11. state<GNAME> – geometry state